Abstract
Loeys–Dietz syndrome (LDS) is caused by connective tissue mutations; the resulting defective connective tissue in organs such as the eye may be related to ocular symptoms in patients with LDS. The aim of this study was to review different ocular manifestations in LDS. A literature review of articles published within the past 5 years was performed using Web of Science™ and PubMed to search for ‘Loeys–Dietz’ with the terms ‘ocular’ and ‘ophthalmology.’ Additional search terms were generated from the initial literature assessment, and 32 articles were ultimately reviewed. Reported ocular symptoms in LDS included hypertelorism, ocular misalignment, refractive errors, and more. For LDS, the most reported findings were hypertelorism (n=111), astigmatism (n=25), down slanting palpebral fissures (n=20), myopia (n=9), and strabismus (n=8). However, more research on ocular symptoms in LDS is needed.
Key Points
1. Studying the presence of ocular symptoms in Loeys–Dietz syndrome is important because of how it affects patient management.2. The most referenced ocular findings for Loeys–Dietz syndrome included hypertelorism, astigmatism, and down slanting palpebral fissures.
3. A large database of the ocular involvement of these conditions could facilitate earlier detection and potential treatments.
INTRODUCTION
Loeys–Dietz syndrome (LDS) is a rare disorder of connective tissue, which classically presents with signs of hypertelorism, bifid uvula, aneurysms, or arterial tortuosity.1 Previous categorisation of the LDS subtypes considered the phenotype to be strongly correlated with subtype; for example, craniofacial features were thought to be strongly associated with LDS Type I, and osteoarthritic features strongly associated with LDS Type III. There has since been a shift in thinking, such that each LDS subtype is now considered to be a combination of features.2
There are six types of LDS, caused by mutations in TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, and SMAD2, respectively.3 These genes are involved in the transforming growth factor-β pathway, which is critical for tissue development, differentiation, and maintenance.4 Both TGFBR1 and TGFBR2 code for receptors in the pathway, which are acted on by cytokines coded by TGFB2 and TGFB3, while SMAD2 and SMAD3 help to mediate the signals of this pathway.3 Ocular embryogenesis begins with the development of the optic vesicles, which are influenced by several signalling molecules, including the transforming growth factor-β.5 Thus, it follows that there might be ocular abnormalities in patients with LDS. This study sought to review the ocular manifestations of LDS, as well as their relative frequencies.
METHODS
A literature review was performed using Web of Science™ and PubMed. Eligibility criteria included whether the studies were published within the past 5 years and written in English, however, all types of articles were included due to the niche topic area. Initially, the terms ‘Loeys–Dietz, ‘ophthalmology’, and ‘Loeys–Dietz, ‘ocular’ were searched; more specific search terms were then generated from the articles generated in the initial search. Those additional terms used were: ‘eye’, ‘retina’, ‘retinal detachment’, ‘xerophthalmia’, ‘lenticular’, ‘lens opacity’, ‘glaucoma’, ‘myopia’, ‘canthal’, ‘canthal rhytids’, ‘sclera’, ‘scleral fragility’, ‘ocular fragility’, ‘microcornea’, ‘angioid streaks’, ‘vitreous’, ‘brittle cornea’, ‘globe perforation’, ‘keratoconus’, ‘keratoglobus’, ‘refractive error’, ‘cornea’, ‘corneal fragility’, ‘corneal rupture’, ‘corneal hydrops’, ‘lens subluxation’, ‘conjunctivochalasis’, ‘Bruch’s membrane’, ‘choroidal neovascularisation’, ‘exotropia’, ‘central corneal thickness’, ‘ectopia lentis’, ‘cataract’, ‘hypertelorism’, ‘craniofacial’, and ‘astigmatism’.
The titles, abstracts, and full texts were reviewed for each potential article. The internal validity of each study was rated by two reviewers. An initial evaluation of internal validity was assessed using the National Institutes of Health (NIH) Study Quality Assessment Tool. However, due to the large number of case reports in this study and the inability to find a broadly appropriate category to assess these reports, the Joanna Briggs Institute (JBI) method was ultimately used.
RESULTS
Loeys–Dietz Syndrome
After the exclusion criteria were applied, there were 32 appropriate articles, which described a total of 275 patients (Table 1).
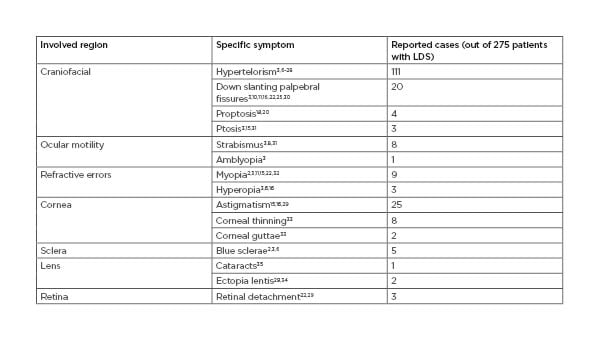
Table 1: A summary of the number of reports of various ophthalmic symptoms in patients with Loeys–Dietz syndrome from literature published in the past 5 years.2,3,6-34
LDS: Loeys–Dietz syndrome.
Craniofacial
Hypertelorism is commonly reported in patients with LDS.6-28 It was documented in 111 out of 275 patients in the articles that the authors reviewed. However, one study assessing ocular complications in patients with LDS versus controls found that interpupillary distances were similar in the eyes of the patients with LDS and the control patients. This is interesting because hypertelorism is considered to be a common clinical finding associated with LDS. However, no specific measurements were noted in the original report describing this phenotype.29
Down slanting palpebral fissures3,15,18 and proptosis were also reported in patients with LDS.3,10,11,16,20,22,25,30 Down slanting palpebral fissures were documented in 20 out of 275 patients, while proptosis was documented in four out of 275 patients.
Eyelid
Eyelid ptosis has also been described in patients with LDS.3,15,31 A total of three out of 275 patients with LDS were affected in the articles reviewed.
Ocular Motility
LDS has been linked to features of ocular misalignment, including strabismus3,8,16,22,27,29,31 and amblyopia.29 Of the articles reviewed, eight out of 275 patients had strabismus, one of which also had amblyopia on exam.
Refractive Errors
Near-sightedness has been reported as part of the ophthalmic characteristics associated with LDS.2,3,11,15,22,32 One study found that the eyes of patients with LDS were on average more myopic than the eyes of control patients, although not significantly more so.29 Of the studies reviewed, nine out of 275 patients with LDS had documented myopia. Hyperopia has also been described in patients with LDS3,6,16 and was found in three out of 275 patients.
Sclera
Blue sclerae have been linked to LDS.2,3,6,9,33 In total, blue sclerae were documented in five out of 275 patients with LDS.
Cornea
Keratoconus has been associated with LDS.29,35 Of the articles reviewed, two out of 275 patients with LDS were affected. In one study, although not significantly so, the eyes of patients with LDS had thinner central corneas on average than those of the control patients.29 Decreased corneal thickness was also found in all three patients in an LDS case report;34 the authors reference previous research that reported an average difference in thickness of 21 µm between patients with LDS mutations and control patients. This is important to study because decreased central corneal thickness is a risk factor for primary open angle glaucoma.33 In another study, there were three patients with corneal thinning, two of which also had corneal guttae.34
Astigmatism has also been described in patients with LDS.15,16,29 Reports of astigmatism were found in 25 out of 275 patients in the articles reviewed.
Lens
Pseudophakia was found more commonly in the eyes of patients with LDS versus those of control patients (4.0% versus 0.8%) in one study,29 although this may have been a coincidental finding considering the high incidence of cataracts in the general population. Lenticular opacities have also been reported in patients with LDS22 and were noted in one out of 275 patients.
Some studies reported patients with LDS with ectopia lentis.29,33 A total of two out of 275 patients with LDS had documented ectopia lentis. However, other studies did not find any cases in patients with LDS.15,20
Retina
Retinal detachments can occur in patients with LDS22,29 and were described in three out of 275 patients with LDS in total. Two of these patients were diagnosed with familial exudative vitreoretinopathy (FEVR).22 The prevalence of FEVR in LDS is uncertain, possibly because it is more likely to be missed than other ocular pathologies. However, the authors propose that the findings of both FEVR and aortic dilation may suggest LDS. Early FEVR identification is important to prevent progression of neovascularisation and blindness.22
Miscellaneous
Abnormality of the macular reflex was described in one patient.2 Another patient experienced ptosis and anisocoria due to Horner syndrome, which was secondary to a subclavian artery aneurysm leak after stent graft placement.36
DISCUSSION
Studying the presence of ocular symptoms in LDS is important because of how it affects patient management. Previous studies have suggested a relationship between craniofacial abnormalities, including hypertelorism, and vascular events in patients with LDS, with more severe features being related to the first vascular event occurring at an earlier age.24
The most referenced ocular findings for LDS included hypertelorism (n=111), astigmatism (n=25), down slanting palpebral fissures (n=20), myopia (n=9), and strabismus (n=8). Interestingly, in one study, the eyes of patients with LDS had fewer cases of the following ocular issues compared to control patients: glaucoma, post-retinal detachment repair aphakia, cataracts, and iris transillumination defects.29
One limitation of this study was the small amount of available data. Connective tissue disorders can sometimes go undiagnosed; ophthalmic symptoms may not always be at the forefront of a patient’s diagnosis or treatment due to their lower mortality risk. Also, some researchers may have difficulty attributing ocular symptoms to a disease process rather than genetic predisposition. For example, myopia is commonly found in patients with LDS, but it is also a very prevalent condition for healthy people as well, occurring in approximately 23% of the world population.37 As a result, ocular symptoms may not be a focus in patient documentation and, in turn, less data are available for publishing.
Another limitation of this study was that not all articles had a specific prevalence for ocular manifestations. For example, one article reported hypertelorism in a category that also included other craniofacial features, such as cleft palate. As it was not possible to determine how many patients specifically had each symptom, this result was excluded from the frequency count. This may have led to the underreporting or overreporting of certain ocular symptoms.
An additional limitation of this study is related to the unstandardised measurement of certain ocular symptoms. Scleral discolouration and drooping eyelids are relatively subjective to diagnose. As such, there may be a bias in the recording of these symptoms based on whether a patient has a connective tissue diagnosis already and whether these symptoms are being ‘looked for’. Although hypertelorism can be measured, most of the articles reviewed did not report measurements against a standard number to define a patient as having hyper- or hypotelorism.
Future research could include a longitudinal, multicentre study that records ophthalmic conditions in patients with LDS prior to and after diagnosis. Having a large database of the ocular involvement of these conditions could promote earlier detection and potential treatments. Additionally, it may improve the understanding of how ophthalmic features, in addition to craniofacial features, may be related to vascular complications in these patients.