Abstract
Ageing is a complex process of accumulation of molecular, cellular, and organ damage, leading to loss of function and increased vulnerability to disease and death, the rate and extent of which varies among individuals. Osteoarthritis (OA) is not only the most common joint disease, but is also one of the major causes of disability in people aged >65 years and is accompanied by comorbid conditions, increased mortality, and decreased quality of life. One of the major risk factors for OA is ageing. However, OA itself may be involved in the biological ageing process. This is likely to be in part a direct involvement, by contributing levels of systemic inflammation and sharing molecular pathways with biological ageing, such as mitochondrial damage leading to cell senescence. Although OA is not considered an inflammatory form of arthritis, there is evidence of subclinical low-grade inflammation in the whole joint and inflammatory processes play a key role in the disease pathogenesis. For instance, there is synovial inflammation (e.g., following injury), mechanically derived inflammation present due to biomechanical overloading of a joint, and systemic inflammation resulting from obesity. Systemic inflammation is often associated with frailty, and having a high concentration of inflammatory markers is predictive of incident frailty, some of which are known to increase with age and correlate with pain. In addition, OA may also contribute indirectly to biological ageing via the disability and pain resulting from it. Further research into the exact process linking OA and biological ageing, including frailty, is needed.
INTRODUCTION
Osteoarthritis (OA) is the most common global chronic joint disease. The disease may affect single or multiple joints and even be generalised. OA is a chronic arthropathy affecting the entire joint, involving the cartilage, joint lining, ligaments, and underlying bone. In OA, cartilage loss, osteophyte formation (bone spurs), and subchondral bone sclerosis leads to pain, disability, and a reduction in quality of life.1 Structural changes, visible by radiography, include narrowing of the joint space, osteophyte formation, and bone remodelling around the joints. OA can arise in any synovial joint in the body but is most common in the large joints (knees and hips), hands, and spine.1
OA is believed to result from both biomechanical and molecular changes in the joint brought about by injury, joint malalignment, obesity, ageing, and inflammation. OA is classified as idiopathic or secondary to anatomic abnormalities, trauma, or inflammatory arthritis. The American College of Rheumatology (ACR) criteria developed for hand, hip, and knee OA are intended to distinguish OA from other causes of symptoms and are best suited to clinical settings in which a high prevalence of other forms of arthritis and joint pain is expected. This is different from the definition used for epidemiological studies given the poor correlation between radiographic disease severity (joint damage) and the joint pain and functional impairment presented by a patient. Thus, OA can be defined pathologically, radiographically, or clinically, but most epidemiological studies have relied upon radiographic features to characterise the disease.2
EPIDEMIOLOGY OF OSTEOARTHRITIS
OA is one of the most disabling diseases in developed countries. Global estimates are that 9.6% of men and 18.0% of women >60 years of age have symptomatic (painful) OA. Eighty percent of patients with OA have limitations in movement and 25% cannot perform their major daily activities.3 World Health Organization (WHO) data also demonstrated that OA moved from the 12th to the 6th leading cause of years lost to disability or morbidity between 2002 and 2007. Increases in life expectancy and ageing populations are expected to make OA the fourth leading cause of disability by the year 2020.3
Individuals with OA (defined both symptomatically and radiographically) at the knee or the hip show a 55% excess in all-cause mortality. Diabetes (95% increased risk), cancer (128% increased risk), cardiovascular disease (38% increased risk), and the presence of walking disability at baseline (48% increased risk) are independently associated with the excess in all-cause mortality.4 Importantly, deaths from cardiovascular causes are higher in patients with walking disability due to OA (72% higher), even after adjustment for baseline covariates, indicating that there is an interplay between the underlying OA and the additional comorbid conditions, which result in a higher risk of mortality. Thus, although the main clinical symptoms of OA are pain and disability, the consequences of the disease are much more far reaching.
Several risk factors that have been recognised to affect hip and knee OA are body weight, age, female sex, occupational activity and injury,2 congenital abnormalities and joint shape, meniscal tears, presence of OA at other joints (Heberden’s and Bouchard’s nodes), and foot and knee alignment, in addition to genetic predisposition. Furthermore, specific inter and intra-articular patterns of OA may represent subsets that have different risk factor profiles and disease courses.
PAIN AND DISABILITY IN OSTEOARTHRITIS
In general, the main symptom of OA is joint pain exacerbated by exercise and relieved by rest, although pain at rest or during the night is not uncommon in advanced disease. Knee pain due to OA is usually bilateral and is experienced in and around the knee. Hip pain due to OA is felt in the groin and anterior or lateral thigh. Hip OA pain can also be referred to the knee. Signs of OA include reduced range of joint movement, joint swelling/synovitis (warmth, effusion, synovial thickening), crepitus, periarticular tenderness, bony swelling, and deformity due to osteophytes.1
OA patients report with a diminished ability to perform the basic activities of daily living, such as climbing stairs or changing from a sitting to standing position.5 In the UK, a recent survey, ‘OA Nation’, found that 81% of people with OA experience constant pain and face limitations in performing certain tasks.4
OSTEOARTHRITIS AND AGEING, MOLECULAR MECHANISMS OF CARTILAGE DEGENERATION
Although late-onset articular cartilage degeneration is common and age is one of the most important risk factors for the disease, the relationship between old age and OA is not fully understood.6 In the past, it was believed that the link with age was due to ‘wear and tear’ of articular cartilage by continuous mechanical stress; we now know, however, that OA involves an active response to injury comprising remodelling of articular cartilage and subchondral bone, in addition to synovial inflammation and damage to other joint structures, such as ligaments and menisci.7
Biological ageing is a complex process and it is now widely accepted that ageing starts with molecular damage, leading to cell, tissue, and, ultimately, organ dysfunction.8 Extensive evidence from animal models and in vitro studies has shown that mitochondria contributes to specific aspects of the ageing process, including cellular senescence, chronic inflammation, and the age-dependent decline in stem cell activity.9
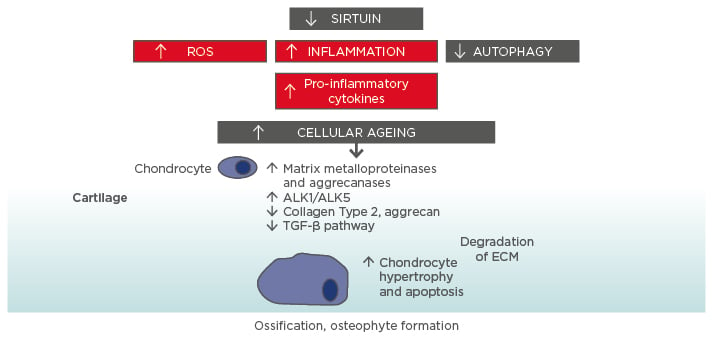
Perhaps the best known and most long-standing hypothesis to explain ageing is the free radical theory, which proposes a central role for the mitochondrion as the principle source of intracellular reactive oxygen species (ROS) leading to mitochondrial DNA (mtDNA) mutations (Figure 1).8,9
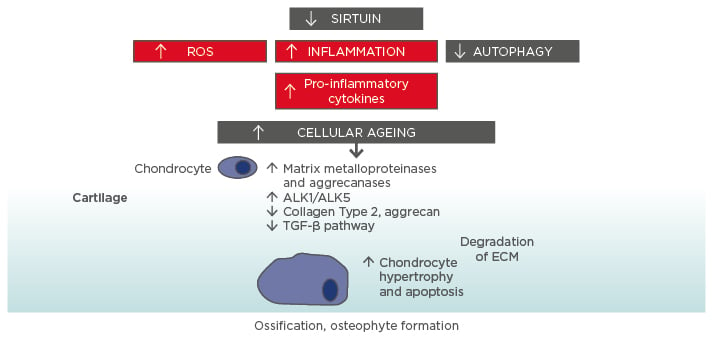
Figure 1: Molecular ageing mechanisms and risk of osteoarthritis.
Various cellular signalling mechanisms, including a decrease in the action of sirtuins in mitochondria, are involved in the process of cellular ageing, resulting in an increase in reactive oxygen species and inflammation, and a decrease in autophagy (lysosome-mediated degradation of damaged proteins and organelles).56-58 These changes influence the expression of catabolic factors resulting in increased production of matrix metalloproteinases, aggrecanases, and pro-inflammatory cytokines, reduced TGF-β signalling, increase in TGF-β receptors ALK1/ALK5 ratio (a cause for elevated metalloproteinases-13 expression in osteoarthritis), and reduced levels of collagen Type 2 and aggrecan synthesis.59 Mitochondrial dysfunction, inflammation, chondrocyte hypertrophy, and apoptosis all contribute to the development of osteoarthritis, with the resulting formation of osteophytes.
ALK: activin receptor-like kinase; ECM: extracellular matrix; ROS: reactive oxygen species; TGF: transforming growth factor.
Over the past 10 years, substantial evidence has accumulated showing that differences in mtDNA haplogroups correspond to variations in prevalence and progression of cartilage loss in large joint OA.10 The mtDNA haplotypes T, J, and the JT cluster, on the other hand, are significantly associated in populations from the USA, the Netherlands, and Spain, with radiographic incidence and progression of the disease.11 Fernandez-Moreno et al.11 report that the mtDNA haplogroup J, the same haplogroup associated with lower OA prevalence, lower disease progression, and lower cartilage loss, is also associated with a significantly lower risk of incident knee OA.
The functional relevance of these mitochondrial haplotypes has been recently shown, using cytoplasmic hybrid (cybrid) cell lines.10 Cybrids incorporate mitochondria from human subjects and perpetuate the mtDNA-encoded components while maintaining the nuclear background of different cybrid lines as constant;12 thus, allowing investigators to assess the influence of mtDNA variation on cell function.
The cybrids carrying the haplogroup H produce higher adenosine triphosphate levels than those with the haplogroup J, but this higher energetic efficiency was accompanied by higher production of ROS and the proportion of cells that survived in the presence of hydrogen peroxide was almost half the number of cybrids with haplogroup J. In chondrocytes during OA, oxidative stress may act together with inflammatory and/or mechanical stress to accentuate catabolic processes by increasing the levels of ROS relative to antioxidants.13 The increased levels of ROS also contribute to the senescence secretory phenotype, in which the age-related decline in the responses of chondrocytes to anabolic growth factors is related to increased oxidative stress.14 The depletion of antioxidants promotes mitochondrial dysfunction in chondrocytes,15 which, in turn, can amplify the stress responses through increased production of nitric oxide and ROS and NF-κB signalling.15,16
These data, therefore, prove the functional relevance of mtDNA variation linked to risk of OA on cell function and survival and is in agreement with recent work by the same group showing that OA cartilage presents signs of early molecular ageing compared to healthy age-matched cartilage.6
This increase in cartilage ageing may also reflect overall higher ageing in other organ systems and may be related to the higher rate of comorbidities seen in OA patients.17 Indeed, OA is also linked to biological ageing overall and strong links have been found between OA and frailty. An overview of some of the molecular mechanisms underlying the connection between cellular ageing and cartilage damage is presented in Figure 1.
OSTEOARTHRITIS AND SYSTEMIC INFLAMMATION
Although OA is not considered an inflammatory form of arthritis, there is evidence of subclinical low-grade inflammation in the whole joint and inflammatory processes play a key role in the disease pathogenesis.18 Synovitis (inflammation of the synovium) is a critical characteristic of OA and is often considered the driver of the OA process. However, inflammatory processes are initiated via mediators that are released not just by the synovium but by bone and cartilage too.18
The drivers of this inflammation are varied and several sources of inflammation are involved in OA pathogenesis. In the first instance, there is synovial inflammation, which also affects cartilage and bone. Following a traumatic injury or a repetitive micro trauma to a joint, fragmented cartilage within a joint space provokes a reaction from synovial cells. As the fragments are considered foreign bodies, the synovial cells release inflammatory mediators. These mediators activate chondrocytes and produce metalloproteinases (MMP), which promote cartilage destruction. These mediators also induce inflammatory cytokines and MMP by the synovium itself that perpetuates further destruction of the synovium. Altered rates of osteophyte remodelling in OA are due to increased or decreased osteoclastic bone resorption.19 There is evidence in the literature of chemical communication between chondrocytes and osteophytes.20 Inflammatory mediators such as cytokines can move between these tissues, consistent with the fact that OA is a disease of the whole joint. Moreover, mechanical inflammation is also present due to biomechanical overloading of a joint that is detected by mechanoreceptors located at the joint surface. An abnormal increase in joint loading increases the expression and release of inflammatory cytokines, chemokines, and prostaglandins.21 At an intracellular level, the mechanical forces are translated into chemical signals, also known as mechanotransduction, that trigger inflammation and gradual onset of OA. The presence of atherosclerosis-related inflammation and altered levels of adipokines offer a further contribution to systemic inflammatory processes in addition to joint-localised ones in OA.
It is known that obesity induces an inflammatory environment, because adipose tissues express cytokines and adipocytokines that have been identified in the plasma and synovial fluid of OA patients.22-24 Obesity can also accelerate the OA process by inducing ischaemia at the subchondral level. The direct ischaemic effects on the bone are known to reduce cartilage nutrition and inflict multiple bone infarcts that are characteristic of advanced OA. Bone and cartilage remnants can be identified in the synovial and capsule space, with marked eburnation of bone surrounding the infarct. Adipokines also induce insulin resistance, endothelial dysfunction, and a systemic inflammation, which are implicated in atherosclerosis and could explain why OA patients have an almost 40% increased risk of cardiovascular disease.22 Adipokines play an important role in the pathogenesis of OA and have been shown to contribute to the formation of osteophytes.23,24 To date, the best-studied adipokines are adiponectin, leptin, visfatin, and resistin,25 and in animal models it has been shown that a local knee injection of visfatin inhibitor protects mice from developing mechanical OA.26
Inflammation in OA is also involved when an increase of inflammatory mediators means that a increased concentration of oxidised proteins accumulate within the joint. These ROS cause oxidative damage, which can trigger inflammation, promote cell senescence or ageing, and in particular, chondrocyte ageing (Figure 1). In ageing, there is a loss in the ability of cells and tissues to maintain homeostasis, particularly when placed under abnormal stresses such as oxidative stress or biomechanical overloading of a joint. This promotes stress-induced senescence of chondrocytes. Ageing also involves the formation of advanced glycation end-products (AGE), which are produced in ageing tissues. These end-products alter the mechanical properties of cartilage, which leads to more brittle tissue with an increased fatigue failure, and stimulate the overproduction of pro-inflammatory cytokines and MMP.27 Overall, the integrity of the joint structure is compromised internally by inflammatory processes and externally by the systemic effects of ageing and mechanical loading.18 Therefore, while there appears to be a complex interplay between mediators and inflammatory processes affecting different joint structures, the overall effect over time is one of gradual degradation of tissues, loss of optimal joint integrity and function, and the onset of pain (Figure 2).
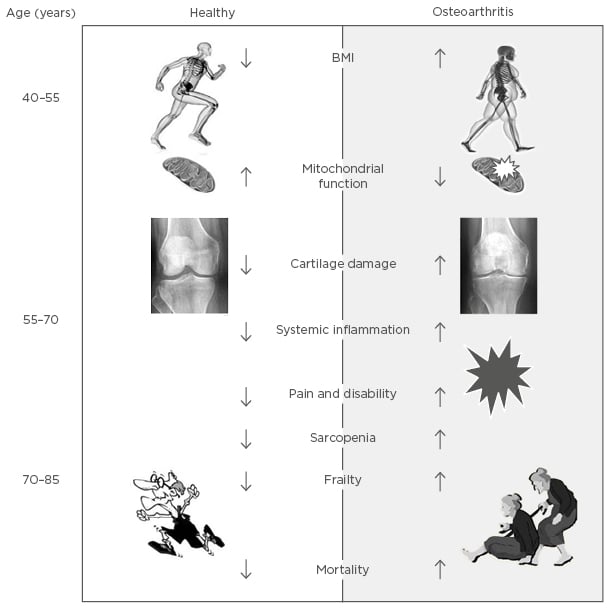
Figure 2: Schematic representation of the link between osteoarthritis and biological ageing.
OSTEOARTHRITIS AND FRAILTY
Knee OA is not only a leading cause of functional limitation and disability in ageing adults, as described above,28 it is also strongly associated with another geriatric condition: frailty.29-31 Clinical frailty is considered highly prevalent in old age and predictive of a high risk of falls, worsening mobility, disability, hospitalisation, and death.32,33 It is an increasing global public health burden with the reported prevalence of frailty in older people varying from between 5.6% in Germany29 to 42.6% in Chile.34
Knee OA and frailty are reported to share common risk factors such as obesity,35,36 with knee OA also associated with greater prevalence of developing frailty.30 Fried et al.32 proposed the definition of frailty as being when ≥3 of the following criteria were present: unintentional weight loss, self-reported exhaustion, grip strength muscle weakness, slow walking speed, and low physical activity. Patients with the presence of one or two frailty criteria are categorised as being in a pre-frail state. Castell et al.29 analysed older adults in six European countries and found the odds of pre-frailty and frailty were 1.54 and 2.96-times higher among OA than non-OA patients, respectively.
Patients with knee OA have been reported to have slow walking speed,37 fatigue,38 low physical activity,39 whilst patients with hip OA have been reported to display all frailty criteria,40 suggesting that patients with OA often display frailty criteria. Unintentional weight loss is often not a characteristic of knee OA; on the contrary a large proportion of severe knee OA is attributable to obesity.41 The underlying mechanisms associating knee OA and frailty are not yet understood, but their identification would provide novel targets for the management and prevention of frailty in adults.
INFLAMMATION AS A POTENTIAL MECHANISM LINKING OSTEOARTHRITIS, FRAILTY, AND SARCOPENIA
In older patients with OA there are positive relationships between pain, frailty, and sarcopenia (Figure 2), although the mechanisms behind the associations are not yet fully understood. In these aged patients, quadriceps muscle weakness has been reported to be present without knee pain, suggesting that the quadriceps weakness is a possible primary risk factor for knee pain and progression of joint damage in people with knee OA.42
OA-related knee pain, along with quadriceps muscle weakness, can lead to a decrease in physical activity.43 This inactivity may lead to the loss of muscle mass; however, systemic inflammation has also been suggested as a potential mechanism that associates OA and frailty.30 Pain itself is a risk factor for the development of frailty. A recent prospective analysis, of 1,152 non-frail subjects at baseline, has demonstrated that lower limb OA-related pain was associated with an increased risk of developing frailty over 4.4 years, compared with people with OA and no pain.44
Age-related inflammation may not directly cause OA (as presented above) but is likely to act as contributing factors to its development and progression, as well as to increased pain and reduced physical function.45 Obesity associated with OA and ageing further contributes to the inflammatory environment, leading to muscle atrophy and sarcopenia through the production of high levels of inflammatory markers and cytokines, such as C-reactive protein (CRP), tumour necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1-β.46 TNF also contributes indirectly to sarcopenia by causing insulin resistance.
Both sarcopenia and obesity are independently associated with physical disability in elderly people. Sarcopenic obesity (age-related muscle atrophy in obese individuals) results in more physical limitations than sarcopenia or obesity alone, and has been strongly implicated in both risk of OA and frailty.47,48 Systemic inflammation is often associated with frailty; for example, having a high concentration of the inflammatory markers CRP and fibrinogen were predictive of incident frailty in women.49 Higher levels of CRP have also been reported in women with early knee OA, as well as predicting those whose disease will progress over 4 years.50
There have been further systemic markers of inflammation, whose levels in the blood have been demonstrated to increase with age and correlate with pain. TNF-α, along with CRP, has been reported to be positively associated with knee pain over a 5-year study of older adults with OA,51 whilst higher serum levels of IL-6 are also considered to be associated with pain in early-stage knee OA.52 Significant positive relationships have previously been identified between frailty and IL-6,53 with a link also being reported between elevated IL-6 and the loss of bone and muscle.54
The exact mechanism driving the relationship between inflammatory mediators, pain, OA, frailty, and sarcopenia is currently unknown. Inflammatory cytokines involved in frailty, including IL-1, IL-6, and TNF-α, are also increased in OA cartilage as opposed to normal cartilage.55 It is thought that a complex network of inflammatory cytokines including TNF-α, IL-1-β, and IL-10 are involved in the regulation of IL-6 and inflammation.54 Further research into the exact process is needed.
CONCLUSION
OA is the most common joint disease and is highly prevalent after 60 years of age. However, the presence of OA appears to also influence the risk of unhealthy ageing, both as the presence of cardiometabolic comorbidities and as a risk factor for the development of frailty and sarcopenia later in life (summarised in Figure 2). In this article, we have covered some of the evidence linking OA and OA pain to systemic inflammation, development frailty, and sarcopenia. Prospective studies directly linking the systemic inflammation that accompanies OA and pain in OA to the development of frailty and sarcopenia are not yet available. However, the evidence in the literature strongly suggests a functional link, and studies investigating the causal relationship between the two, the molecular mechanisms underlying them, and potential interventions, deserve careful consideration.