Abstract
This observational study aimed to evaluate the efficacy and real-world experience of apremilast (APR) in treating psoriatic arthritis (PsA) patients with co-existing conditions presenting to clinic. Data from 28 patients treated with APR for PsA were collected between January 2016 and January 2019. Outcome measures disease activity score 44-C-reactive protein (DAS44-CRP), 0–68 for tender and 0–66 for swollen joint count, were collected at Weeks 0, 16, and 52. Response was classified using the Psoriatic Arthritis Response Criteria (PsARC). Adverse events or worsening of pre-existing conditions were recorded. Results included outcomes at Weeks 16 and 52 which showed a percentage reduction in mean DAS44-CRP at Weeks 16 and 52 by -1.4 and -1.9, respectively. There was percentage reduction at Weeks 16 and 52 of tender (-55.5%, -75.4%) and swollen (-45.8%, -61.5%) joint counts from baseline. It was also found that 19/28 (68.0%) patients were responders by PsARC criteria up to Week 52. Responders had shorter disease duration (mean: 4.9 years, standard deviation: 1.9) and lower previous exposure to biologic disease-modifying antirheumatic drugs (bDMARD); 16/19 subjects (84.0%) had no previous bDMARD. There were no serious adverse events during the study and no worsening of co-existing conditions during treatment. In this real-world observational study, APR was shown to be effective in PsA patients with multiple co-existing conditions. APR was more effective in PsA patients with shorter disease duration and in bDMARD naïve patients. APR provides another effective treatment option for PsA patients with multiple co-existing conditions.
Introduction
Apremilast (APR) is a small molecule phosphodiesterase-4 inhibitor that is used in the treatment of psoriasis (PsO) and psoriatic arthritis (PsA). Randomised controlled trials (RCT) have shown that APR is effective in both psoriasis and PsA.1,2 The Psoriatic Arthritis Long-term Assessment of Clinical Efficacy (PALACE) RCT have shown APR to be effective in treating PsA.3-6 There is, however, limited real-world data on the use of APR in PsA.7 The objective of this study is to report on the real-world experience and outcomes of using APR in PsA patients with co-existing conditions.
Methods
The authors performed an observational study on the effectiveness and tolerability of APR at a standard dose of 30 mg twice a day, following a loading dose in patients with PsA. All subjects fulfilled classification criteria for PsA (CASPAR)8 and had active disease according to the National Institute of Clinical Excellence (NICE) criteria for treatment with APR in PsA.9 Patients meeting the NICE criteria who were deemed suitable by the attending rheumatologist were commenced on APR. This study was an evaluation of standard clinical practice and outcome of APR use against NICE criteria and, as such, ethical approval was not required.
As part of the NICE guidance, PsA patients were assessed at baseline, at 16 weeks, and every 6 months. Clinical assessments at each visit included the disease activity score 44-C-reactive protein (DAS44-CRP), scoring for tender joints (0–68), swollen joint count (0–66), CRP levels, and patient and physician global assessments on a 5-point Likert scale. High disease activity is defined as a DAS44 of >3.7, moderate activity is defined as a DAS44 between 2.4 and 3.7, low activity is defined as a DAS44 between ≤2.4 and ≥1.6, and remission is defined as a DAS44 <1.6. Efficacy outcomes were recorded at the assessment at Weeks 16 and 52 and compared with the baseline evaluation. The PsA response criteria (PsARC) was calculated and subjects were defined as responders if there was an improvement in at least two of the four PsARC criteria (including joint tenderness or swelling score) with no worsening in any criteria.10 Subjects were classified as responders and non-responders based on the PsARC status up to Week 52. In non-responders who stopped APR at Week 16, this was the last observation measured. Patients who stopped APR between Weeks 16 and 52 were excluded from further analysis at Week 52.
Statistical Analysis
Continuous variables were expressed as median (range) or mean (standard deviation [SD]) as appropriate and binomial variables were expressed as number and percentage. The data followed a normal distribution and comparisons between baseline and follow-up measurements were performed using paired Student’s t-test with significance of the difference set at p<0.05. Significant differences between responders and non-responders were defined as those at a level of p<0.05 by either Student’s t-test or Chi-squared test.
Results
A total of 28 PsA patients (n=16 [57.1%] female, n=12 [42.9%] male) attending the rheumatology clinic between January 2016 and January 2019, who were deemed suitable for APR based on NICE criteria, were included in this study. The patients started on APR within this study had active PsA (>3 tender and >3 swollen joints) and with previous failure to a minimum of two conventional synthetic (cs) disease-modifying antirheumatic drugs (DMARD). The mean age was 53 (SD: 11) years and the mean PsA disease duration 5.9 (SD: 2.5) years. Mean number of csDMARD pre-APR was three. Of the patients in this study, 11 (40.0%) were biologic (b)DMARD inadequate responders prior to commencing APR. No patients had targeted synthetic DMARD prior to commencing APR. Clinical characteristics are shown in Table 1.
The mean DAS44-CRP was measured at baseline and at Weeks 16 and 52. The baseline DAS44-CRP was 3.8 (SD: 0.7). Patients had active PsA with the mean DAS44-CRP >3.7 at baseline. The mean DAS44-CRP was reduced at Weeks 16 (-1.4, SD: 1.0) and 52 (-1.9, SD: 1.1) (Figure 1). The difference in DAS44-CRP at Weeks 16 and 52 compared to baseline was statistically significant p<0.01. Four patients stopped APR at Week 16 due to inefficacy. Two patients stopped APR between Weeks 16–52 (mean: 31.9, SD: 3 weeks). The tender and swollen joint counts were recorded at baseline and at follow up. There was a percentage reduction at Weeks 16 and 52 of mean tender (-55.5% and -69.3%) and mean swollen (-45.8% and -55.1%) joint counts (Figure 2). The reduction in joint counts at Weeks 16 and 52 compared to baseline was clinically significant and reached statistical significance of p<0.01.
Based on the PsARC, 19 of 28 (68.0%) patients were classified as responders and 9 (32.0%) as non-responders up to Week 52 of this study. Subjects that stopped APR during this study were classified as non-responders. Responders had a shorter disease duration (mean: 4.9, SD: 1.9 years) compared to non-responders (mean: 7.3, SD: 2.3 years). This was statistically significant at p<0.05. All patients had prior exposure to csDMARD. Responders had lower exposure to bDMARD with 16 of 19 (84.2%) responders being bDMARD naïve compared to non-responders with 8 of 9 (88.9%) being bDMARD experienced. This association was statistically significant (p<0.01). There was no significant difference in age: responders had a mean age of 50.7 (SD: 12 years)and non-responders had a mean age of 55.4 (SD: 8.5 years; p=0.48).
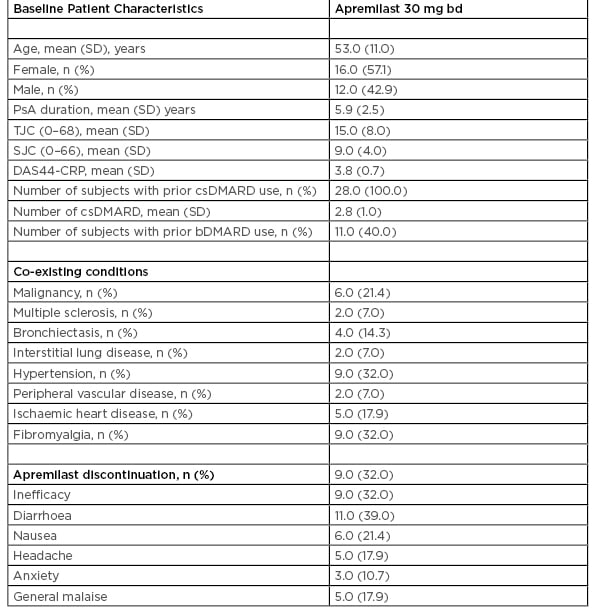
Table 1: Baseline patient characteristic of 28 psoriatic arthritis patients treated with apremilast.
bd: twice daily; bDMARD: biologic disease-modifying antirheumatic drugs; csDMARD: conventional synthetic disease-modifying antirheumatic drug; DAAS44-CRP: disease activity score 44-C-reactive protein; PsA: psoriatic arthritis; SD: standard deviation; SJC: swollen joint count; TJC: tender joint count.
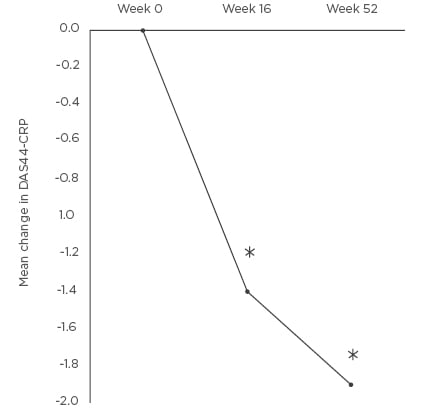
Figure 1: Mean change in disease activity score 44-C-reactive protein at Weeks 16 and 52 compared to baseline.
*p<0.01 DAS44-CRP: disease activity score 44-C-reactive protein.
There was also no significant difference in sex: 7 responders were male (37.0%) and 12 were female (63.0%), and non-responders were 5 males (56.0%) and 4 females (44.0%); p=0.35 was found between the responder and non-responder groups.
Comorbidity is defined as any distinct additional entity that has existed or may occur during the clinical course of a patient who has the index disease under study.11 In this study the prevalence of co-existing and comorbid conditions (Table 1) was included, including previous malignancy (6 patients, 21.4%). The types of cancer included breast (3 patients), lymphoma (1 patient), tongue (1 patient), and kidney (1 patient). The other co-existing conditions presented were multiple sclerosis (2 patients, 7.0%), bronchiectasis (4 patients, 14.3%), interstitial lung disease (2 patients, 7.0%), hypertension (9 patients, 32.0%), peripheral vascular disease (2 patients, 7.0%), ischaemic heart disease (5 patients, 17.9%), and fibromyalgia (9 patients, 32.0%). There was no worsening of co-existing conditions or any serious adverse events while on APR during the study up to 52 weeks. There was also no appreciable improvement in the comorbidities, such as cardiovascular disease or hypertension, during this study.
Of the 28 patients, 9 (32.0%) were discontinued after a mean treatment period of 7.3 months (SD: 3.6) due to lack of efficacy. In the first 52 weeks of APR treatment, the most common side effects were diarrhoea (11 patients, 39.0%) and nausea (6 patients, 21.0%). The gastrointestinal side effect did not necessitate stopping the medication. The other side effects were headache (5 patients, 17.9%), anxiety (3 patients, 10.7%), and general malaise (5 patients, 17.9%). None of these side effects necessitated stopping APR.
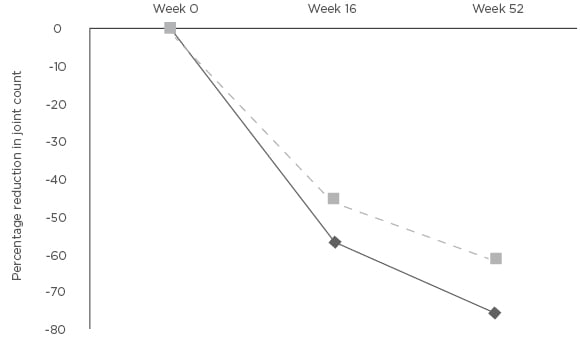
Figure 2: Mean percentage change in tender joint count and swollen joint count at Weeks 16 and 52 compared to baseline.
*p<0.01 SJC: swollen joint count; TJC: tender joint count.
Discussion
Previously published RCT have shown that APR is effective in patients with PsA.3-6 The RCT have also shown an acceptable safety profile of APR in the treatment of PsA. This study reports the real-world experience of the use of APR in unselected PsA patients with active disease. The presence of co-existing conditions in this patient cohort may mean that they could be excluded from clinical trials. There is limited real world data of APR use in PsA7,12 It is therefore necessary to understand the real-world clinical experience of treating unselected PsA patients with multiple co-existing conditions in terms of its efficacy and side effects.
Pooled data from published RCT have shown a reduction in tender and swollen joint counts, as well as DAS28-CRP mean score.3-6 This study also shows the efficacy of APR in treating unselected PsA patients in the clinic. All patients had at least two csDMARD according to the NICE guidance for treatment with APR8 and thus this is applicable to most other clinics which follow this guidance. Patients with shorter duration of PsA and no previous exposure to bDMARD had a better response to APR. The finding of improved response in the bDMARD naïve group is also supported in a RCT.13
The limitations of this study are the small number of patients, lack of controls, and that it is not powered to show a difference in treatments. However, this is a real-world study and patients had a significant clinical improvement. Discontinuation of APR in this study was due to inefficacy based on failure to meet the PsARC target. Gastrointestinal side effects were common but did not necessitate stopping of APR. The other side effects were generally mild and self-limiting. There was no worsening of the underlying co-existing conditions or any serious adverse events during the course of treatment with APR. This is an important real world observation as there is increasing concern about the impact of treatments on co-existing conditions in patients with PsA such as obesity,14 metabolic syndrome,15 and cardiovascular disease.16 Longerterm follow up of these patients will inform the authors as to any benefits of these outcomes.
Conclusion
This study provides real-world clinic experience of the efficacy of APR in the treatment of active unselected PsA in patients with comorbidities. APR was tolerated and the efficacy was established in patients with PsA although they had other co-existing diseases, including cancer. APR improved the clinical outcomes over the first year of treatment compared to baseline. The response to APR was better in patients with shorter PsA duration and in bDMARD-naïve patients. There was an acceptable safety profile with no worsening of the comorbidities during the course of treatment. APR provides an additional treatment option for patients with active PsA. Further studies into its use in patients with multiple co-existing conditions will help determine its place in the treatment pathway for PsA.