Abstract
Psoriatic arthritis (PsA) is a seronegative, inflammatory arthritis associated with cutaneous psoriasis. This disease is associated with significant morbidity, thus requiring early treatment initiation and reduction of disease activity. Anti-cytokine therapies are increasingly being used for the treatment of PsA. In addition to the anti-TNF agents, monoclonal antibodies targeting IL-17 have been approved for the treatment of PsA. Secukinumab is a monoclonal antibody against IL-17 and is currently approved for the management of PsA. In this literature-based review, the current status of secukinumab for the management of PsA is discussed.
Introduction
Psoriatic arthritis (PsA) belongs to the group of diseases known as spondyloarthritis (SpA). The impact of PsA is not limited to just skin and joints, but also includes various extra-articular manifestations and co-morbidities. Asymmetric oligo-arthritis associated with skin psoriasis is the classical presentation of PsA. At present, the Classification Criteria for Psoriatic Arthritis (CASPAR) is used for the diagnosis of PsA.1 With expanding insight into PsA pathogenesis, the roles of various cytokines are becoming more evident, and currently cytokine-targeted therapies are important research topics in the management of PsA. The role of IL-17 in the pathogenesis of PsA has been proven through various studies, and therapies involving monoclonal antibody-targeting of IL-17 are gaining importance in the management of PsA. Secukinumab is a fully human monoclonal antibody that binds to IL-17 and prevents its interaction with the IL-17 receptor. Various clinical trials have shown the beneficial effects of secukinumab in the management of PsA. In this review, the efficacy and safety of secukinumab are discussed in light of currently available research data.
Search Methods
Clinical trials and reviews were searched for using the Pubmed database. The following search terms were used: “Interleukin-17”, or “Secukinumab”, and/or “Psoriatic arthritis”, all of which were selected without time frame or publication date specification. Search items also included efficacy, safety, and radiographic progression (e.g., “Secukinumab efficacy”, “Secukinumab safety” ).
IL-17 in the Pathogenesis of Psoriatic Arthritis
The IL-17 group of cytokines consists of six members: IL-17A–F.2 This recently discovered group of cytokines has led to a paradigm shift in the understanding of SpA pathogenesis. Th-17 cells are important sources of IL-17, and TGF-β1, IL-1β, IL-6, and IL-21 are the polarising cytokines needed by the naïve T cells to transform into Th-17 cells.3,4 Survival of the Th-17 cells is dependent on IL-23.5 In addition to the IL-17 family, various other cytokines are produced by Th-17 cells. Additionally, production of IL-17 is not merely restricted to the Th-17 cells, but also includes γδ T cells, mast cells, neutrophils, ILC 3 cells, and Tc-17 cells.6 It has been shown that synovial fluid from PsA patients contains a large number of IL-17-producing CD4+ T cells compared to patients with osteoarthritis, where both IL-17 and its associated receptor are abundantly expressed.7 An increased number of IL-17-producing CD8+ T cells are also seen in the synovial fluid of PsA patients compared to healthy controls.8 All these observations point towards a strong pathogenic role of IL-17 in PsA and rationale for targeting this cytokine for the management of this disease.
Clinical Trials with Anti-IL-17 Monoclonal Antibody in Psoriatic Arthritis: Secukinumab
Secukinumab is a fully human monoclonal antibody of the IgG1 subclass which binds to and neutralises IL-17, thereby preventing its binding with receptors. IL-17 acts as a pro-inflammatory cytokine. It induces the production of IL-1 and TNF-α. IL-17 promotes osteoclastogenesis by upregulating osteoblast receptor activation by NF-κB ligand. As a result of IL-17 neutralisation by secukinumab, the pro-inflammatory effects of this cytokine are blocked. Secukinumab has a proven efficacy and safety profile for the treatment of PsA.
Efficacy in Psoriatic Arthritis Disease Activity, Skin Score, Functional Status
In a Phase II, proof of concept trial by McInnes et al.,9 secukinumab did not meet the primary endpoint for the American College of Rheumatology 20 (ACR20) response at Week 6 compared to placebo; however, it showed improvement of acute phase reactant levels and quality of life in PsA patients.
The FUTURE 1 study was the first Phase III randomised control trial that showed the efficacy of secukinumab in PsA. In this study, 606 PsA patients were recruited and secukinumab was administered as an intravenous loading dose (LD) of 10 mg/kg at Weeks 0, 2, and 4, followed by subcutaneous injections of either 150 mg or 75 mg every 4 weeks. At Week 24, ACR20 response rates were significantly higher in the secukinumab group compared to the placebo (150 mg [50.0%] and 75 mg [50.5%], placebo [17.3%]; p<0.001 for both comparisons). ACR50 response at Week 24 was also observed in a larger proportion of patients in the secukinumab group compared to the placebo group (150 mg [34.7%] and 75 mg [30.7%], placebo [7.4%]; p<0.001). Significant improvements in Psoriasis Area Severity Index 75 and 90 (PASI 75 and 90) were observed, and sustained efficacy was noted up to 52 weeks. Importantly, 17–19% of secukinumab-treated patients (75 mg and 150 mg groups) were inadequate responders to at least one TNF-inhibitor. Improvement of ACR20 response was observed in these patients also, suggesting the use of IL-17 blockade as an alternative treatment option in these patients.10 In the 3-year extension phase of the FUTURE 1 cohort, persistent efficacy in all endpoints, including ACR20 response, improvement of quality of life, and physical function, were documented in the secukinumab group. ACR20 response at Week 156 was observed in 76.8% and 65.2% of patients from the 150 mg and 75 mg groups, respectively. ACR50/70 responses for these two groups were 54.9/32.9% and 39.0/26.0%, respectively. The ACR20 response was proportionally higher in the anti-TNF naïve patients (81.0% and 67.3% in 150 mg and 75 mg groups, respectively) compared to the previous anti-TNF-experienced patients (61.5% and 55.6% in 150 mg and 75 mg groups, respectively).11 The response rate of secukinumab observed in the FUTURE 1 trial has been summarised in Table 1.
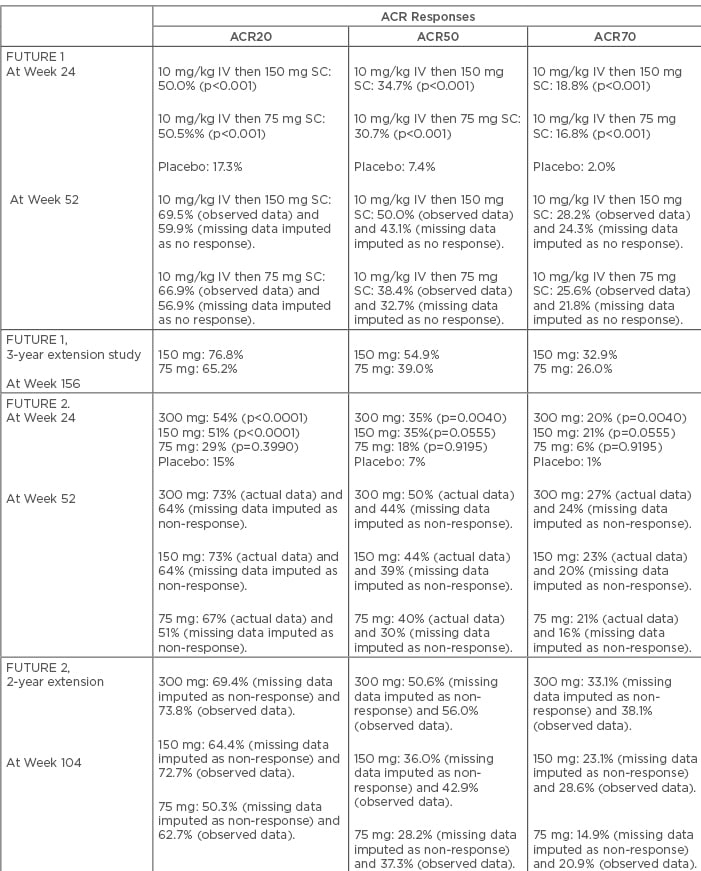
Table 1: Efficacy of secukinumab on the basis of American College of Rheumatology (ACR) responses in the FUTURE 1 and 2 studies.
IV: intravenous; SC: subcutaneous.
FUTURE 2 was a multicentre, placebo-controlled, Phase III trial that included 397 patients with active PsA. In this trial, no intravenous LD was used; instead, subcutaneous secukinumab of 75, 150, or 300 mg or placebo was given at Weeks 0, 1, 2, 3, and 4, followed by the same dose every 4 weeks. Patients of the placebo group were re-randomised based on their response status at Week 16. Non-responders received secukinumab (150 mg or 300 mg every 4 weeks) from Week 16, and responders from Week 24. From an efficacy viewpoint, both 300 mg and 150 mg secukinumab dosages elicited significantly more ACR20 responses compared to placebo (p<0.0001). ACR20 responses at Week 24 were seen in 54%, 51%, and 29% of patients from the 300mg, 150 mg, and 75 mg dosage groups, respectively, compared to 15% of patients from the placebo group. Response rates for the 75 mg secukinumab group were not statistically different from the placebo group (p=0.399), and ACR20 response was sustained through Week 52. ACR50 response was also higher in both secukinumab 300 mg (35%) and 150 mg (35%) groups, compared to the 75 mg (18%) and placebo group (7%). The proportions of patients with inadequate response to at least one TNF inhibitor were 16%, 26%, and 21% in the 300 mg, 150 mg, and 75 mg groups, respectively. Higher ACR response rates were observed in both the anti-TNF naïve and anti-TNF inadequate responders, but response magnitude was higher in the anti-TNF naïve populations. In exploratory analyses, ACR70 response was achieved in 20% and 21% of patients from the 300 mg and 150 mg groups, respectively. These values were higher than the 75 mg (6%) and placebo (1%) groups. Other secondary endpoints, including PASI 75 and PASI 90 response rates and mean changes of DAS28 C-reactive protein from baseline, were significantly higher in the secukinumab 300 mg and 150 mg groups.12 Results from the 2-year extension phase of the FUTURE 2 trial showed persistent improvement of ACR20 response across all three dosage schedules (69.4% in 300 mg, 64.4% in 150 mg, and 50.3% in 75 mg groups). It is important to note that there was sustained increase in the proportion of patients with an ACR20 response over time, not only in the 300 mg and 150 mg groups, but also in the 75 mg group. Sustained clinical response was noted in both anti-TNF naïve and anti-TNF inadequate responders; however, the proportion of patients with ACR20, 50, and 70 rates were higher in the anti-TNF naïve patients. Improvement in functional status and quality of life was also documented.13 Response rates of secukinumab observed in the FUTURE 2 trial has been summarised in Table 1.
The FUTURE 3 trial was designed to find the efficacy and safety of self-administered subcutaneous secukinumab by autoinjector. This study is still ongoing, and the 52-week data has showed significant efficacy of secukinumab as assessed by ACR20 response compared to placebo. More importantly, >99% of patients were able to self-administer the drug at Week 1 successfully. Absence of pain or reaction was reported by >90% of the users, and almost 88% of patients were either satisfied or very satisfied with the use of autoinjector and opined in favour of its user-friendliness.14
The FUTURE 4 study is intended to find out the safety and efficacy of subcutaneous secukinumab 150 mg with or without a LD compared to the placebo. This study is still ongoing, with an abstract having been published at the Pan-American League of Association for Rheumatology (PANLAR) congress in Buenos Aries, Argentina, in 2018. In concordance with the previous trials, it showed significantly higher ACR20 response rates in the secukinumab groups at Week 16 (41.2% in 150 mg, LD; 39.8% in 150 mg, no LD; and 18.4% in placebo; p<0.001 for both secukinumab groups versus placebo). These improvements were sustained up to 52 weeks. Clinical responses were observed in both anti-TNF naïve and inadequate responders; however, the former group showed better responses. PsA patients who received LD showed earlier and better responses than those who did not, and this was mostly observed in the TNF inadequate responders. To summarise, LD of secukinumab may be more effective in PsA patients who have previously shown inadequate response to anti-TNF agents.15
FUTURE 5 is another ongoing trial, aimed at evaluating secukinumab efficacy in reducing symptoms and radiographic progression among PsA patients. In this large study with 996 patients with active PsA, the following three dosage regimens of secukinumab have been used: 300 mg or 150 mg with LD, and 150 mg without LD. ACR20 response at Week 16 were achieved in significantly more proportions of patients in the secukinumab groups compared to the placebo (300 mg with LD [62.6%], 150 mg with LD [55.5%], 150 mg without LD [59.5%], and placebo [27.4%] [p<0.0001 for all]).
This is, however, short-term data, and we must wait for further results to be released.16
Role in Management of Dactylitis
Dactylitis is defined as swelling of an entire digit that is not merely restricted to joints and is frequently seen in patients with SpA. In a recent study with 1,282 PsA patients, dactylitis was identified in 59.2% of patients.17 An earlier Canadian study with 537 PsA patients showed that dactylitis was found in 48.0% of patients.18 Indeed, dactylitis may be present in 29.0–33.5% of PsA patients at first presentation.19 Flexor tendon tenosynovitis and synovitis of joints are the most commonly described pathologies in dactylitis. Soft tissue thickening and extensor tendonitis may also be present. Some studies have demonstrated a link between dactylitis and digital polyenthesitis.20,21
Data from the FUTURE trials showed the beneficial effect of secukinumab in the resolution of dactylitis. In the FUTURE 1 trial, 51.5% of PsA patients of both 150 mg and 75 mg groups had dactylitis. After 24 weeks of treatment, 52.4% of patients had resolution of dactylitis (combined data of both groups), compared to 15.5% in the placebo group.10 This effect was sustained and increased with continuation of secukinumab, and resolution of the condition at Week 156 was seen in 88.1% and 86.8% of patients of 150 mg and 75 mg dosages, respectively.11
The proportion of patients with dactylitis was somewhat less in the FUTURE 2 trial (46%, 32%, and 33% in dosage groups 300 mg, 150 mg, and 75 mg, respectively). Analysis of pooled data regarding resolution of dactylitis across all secukinumab groups did not show any significant difference from placebo (p=0.9195).12
The proportion of patients with dactylitis resolution at Week 104 was much higher than that observed at Week 24. Analysis based on the observed data showed that 88.5% (300 mg group), 92.2% (150 mg group) and 95.6% (75 mg group) of patients were free from dactylitis. These proportions were slightly lower when the analysis was done based on missing values as non-responders (79.9%, 78.0%, and 88.6% for 300 mg, 150 mg, and 75 mg, respectively).13
The Week 52 results from the FUTURE 3 trial showed dactylitis resolution rates of 60.9% and 52.8% for the 300 mg and 150 mg groups, respectively.14 Data from all these studies have clearly documented the efficacy of secukinumab in the management of dactylitis in patients with PsA. It is also obvious that improvement of dactylitis occurs with longer treatment duration. The efficacy of secukinumab in the resolution of dactylitis is presented in Table 2.
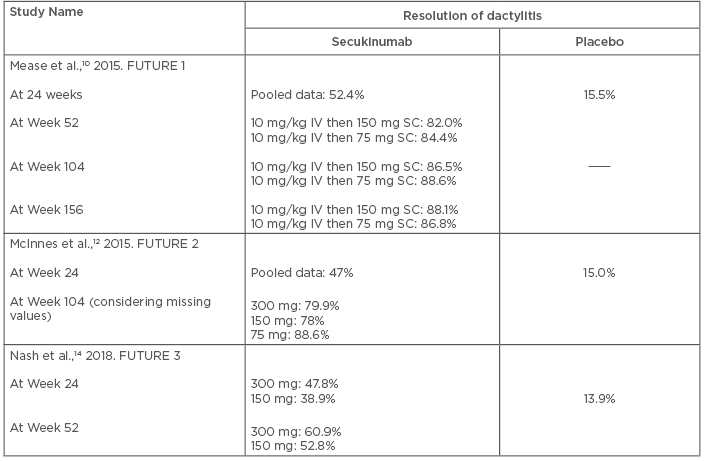
Table 2: Efficacy of secukinumab in the resolution of dactylitis.
IV: intravenous; SC: subcutaneous.
Role in Management of Enthesitis
Enthesitis is defined as inflammation at the sites of tendon and ligament insertion into the bone. In PsA, enthesitis may be a presenting feature or may appear later in the disease course. It has an estimated prevalence of 35.0%, and an annual incidence of 0.9%. Common sites of enthesitis are at the Achilles tendon insertion, plantar fascia, or the lateral epicondyles, but other sites may also be involved.22 The efficacy of secukinumab in the resolution of enthesitis has been documented in previous studies. After 52 weeks of treatment with secukinumab, enthesitis resolution proportion ranges from 46.3–80.3% in different studies.11,13,14 Enthesitis resolution rate increases with longer treatment duration. The efficacy of secukinumab in the resolution of enthesitis has been presented in Table 3.
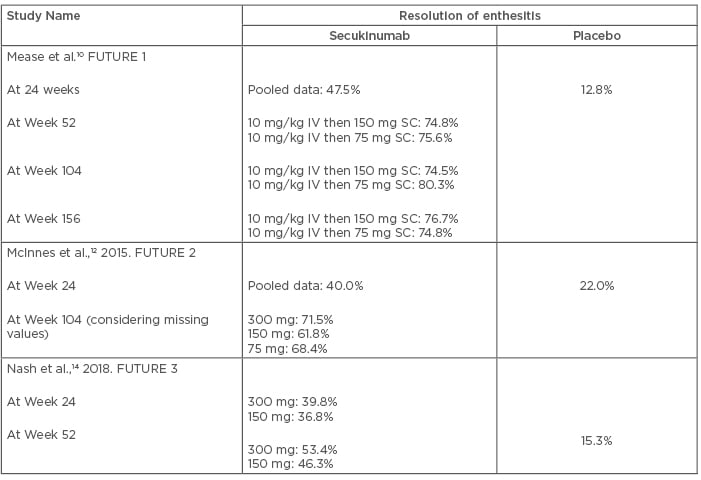
Table 3: Efficacy of secukinumab in the resolution of enthesitis.
IV: intravenous; SC: subcutaneous.
Effect on Radiographic Progression
Retardation of radiographic progression is another important goal in the management of PsA. Both erosion and osteoproliferation are characteristic features of bony changes in PsA. It has been estimated that 12–47% of PsA patients develop bony erosions within 2 years of disease onset.19 The combination of systemic bone loss, along with new bone formation at entheseal and periosteal sites in the SpA group, is possibly mediated by IL-23 and IL-17.23-25 IL-17 promotes osteoclastogenesis thorough a signalling cascade that leads to NFκB ligand expression on osteoblasts, causing their subsequent differentiation into osteoclasts.26,27 IL-17 also has a direct effect on osteoclast differentiation.28 On the contrary, the effect of IL-17 on osteoblasts is yet to be clearly elucidated. Evidence from various studies is both contradictory and supportive regarding the inhibitory effect of IL-17 on osteoblasts.29,30 A recent review concluded that the effect of IL-17 on the osteoblast depends upon multiple factors, including type of cell exposed to IL-17, the differentiation stage of that cell, and the timing and duration of IL-17 exposure.31
The effect of secukinumab on radiographic progression in PsA is quite encouraging. Data from the FUTURE 1 trial showed that patients who received secukinumab had significantly less radiographic progression than placebo group at Week 24, as assessed by mean change of van der Heijde-modified total Sharp score (mTSS) from baseline (0.13 for 150 mg group, 0.02 for 75 mg group, 0.08 for secukinumab pooled doses, and 0.57 for placebo group). At Week 52, mean changes of mTSS from baseline were 0.37, 0.22, and 0.30 for the 150 mg, 75 mg, and pooled dose groups, respectively. Three-year data showed 78.1% (150 mg) and 74.8% (75 mg) patients were radiographic non progressors (non-progression was defined as patients who had a change from baseline of ≤0.5 in mTSS during observation period).11 Sub group analysis did not show any significant difference in the proportion of patients with radiographic nonprogression between anti-TNF naïve and anti-TNF experienced patients. In the secukinumab 150 mg arm, 78.0% (anti-TNF naïve) and 78.6% (anti-TNF experienced) patients were non progressors, whereas in the 75 mg arm these values were 77.7% and 65.5% respectively.11
The recently published primary result of the ongoing FUTURE 5 trial is also in favour of the beneficial effect of secukinumab in radiographic progression in PsA. At Week 24, the mean changes of mTSS from baseline were significantly lower in all secukinumab arms compared to the placebo arm. The proportions of radiographic non-progressors at 24 weeks were 88.0% (300 mg with LD), 79.8% (150 mg with LD), 83.8% (150 mg without LD), and 73.6% (placebo).16 It is notable that significant proportions of patients in the placebo group were also radiographic non-progressors. However, these data were based on a 24-week observation period and radiographic progression was assessed by X-ray. Consequently, significant changes in radiographic score may not be observed within this short time frame. Long-term data of this study may provide additional information regarding this issue.
In previous studies, radiographic progression was assessed by X-ray. It may take a longer time to appreciate significant changes by this imaging technique. MRI and CT can detect bony changes earlier than X-ray. In an open label study by Kampylafka et al.,32 with 20 active PsA patients, changes in the hand joints after 24 weeks of secukinumab treatment were assessed by MRI, power doppler ultrasound, and high-resolution peripheral quantitative CT. They searched for changes in the status of synovitis, periarticular inflammation, bone erosion, enthesiophyte formation, and bone structure. There was no progression in bone erosions or enthesiophytes as detected by MRI and high-resolution peripheral quantitative CT. Secukinumab treatment resulted in cessation of progression of both catabolic and anabolic bone changes in the peripheral joints of PsA patients.32 Data available so far from these studies is clearly indicative of protective effects of secukinumab in the radiographic progression of PsA. Sensitive imaging modalities can detect this effect earlier than through conventional radiography.
Safety Data
For the widespread use of any biologic agent, safety issues are a major concern. Adverse event rates from the FUTURE 1 trial during the 16-week placebo controlled period did not show any significant difference between secukinumab and placebo groups (64.9% in 150 mg, 60.4% in 75 mg, and 58.4% of patients in the placebo group).10 Most of the adverse events were of mild-to-moderate degree. During the 104-week extension periods of the FUTURE 1 and 2 trials, nasopharyngitis was a common adverse event noted in the secukinumab group (among 13.4% and 13.6% patients in FUTURE 1 and 2 trials, respectively), followed by upper respiratory tract infections (among 12.6% of patients in both FUTURE 1 and 2 trials). Diarrhoea and headache were also seen, but less frequently. Oral candidiasis was reported in four patients each in the 150 mg and 75 mg groups (approximately in 2% of patients). No treatment withdrawal was required for candida infection. New tuberculosis or its reactivation was not reported. Neutropaenia was also reported in both FUTURE 1 and 2 trials, but rates of malignancy or inflammatory bowel disease were quite low. In the FUTURE 1 trial, exposure-adjusted rates of serious events among secukinumab 150 mg and 75 mg arms were 11.5 and 7.4 per 100 patient-years, respectively. During the safety period, four patients in the secukinumab 75 mg group had a stroke (exposure-adjusted rate: 0.6/100 patient-years). Two patients from both 150 mg and 75 mg groups experienced myocardial infarction (rate: 0.3/100 patient-years). Drug discontinuation rates were low across all these trials. Drug discontinuation rate in the 156-week extension period of the FUTURE 1 trial were 6.21% and 3.40% in secukinumab 150 mg and 75 mg groups, respectively (data from patients in extension full analysis set).11 Two year data from FUTURE 2 trial showed that 86% and 76% of patients in the 300 mg and 150 mg groups, respectively, completed the 104-week trial period.13
Additional Therapies
In addition to secukinumab, other molecules targeting IL-17 are also under investigation in clinical trials. Ixekizumab is a humanised, monoclonal antibody targeting IL-17. One recent trial evaluated the efficacy of this biological agent in the management of PsA. Initial reports of a Phase III trial with ixekizumab in biologic-naïve PsA patients (SPIRIT-P1) showed the efficacy of this agent in reducing disease activity and improving physical function.33 Fifty-two week data of the SPIRIT-P1 trial also showed sustained efficacy and reasonable safety profile of this drug in PsA.34 Bimekizumab neutralises both IL-17 and IL-17F. One recent proof of concept trial showed its efficacy in PsA.35 Brodalumab is a fully human, recombinant monoclonal antibody of IgG2 subclass which binds to the high affinity receptor IL-17RA. It is already approved for the management of severe plaque psoriasis, and some previous studies have shown its efficacy in the treatment of PsA. However, suicidal tendency may be a major symptom concerning the use of Brodalumab.
Current Recommendations for Use of Secukinumab and Other Biologics in Psoriatic Arthritis
Secukinumab is being approved by the European Medicines Agency (EMA) for the treatment of adult patients with active PsA. As per the European League Against Rheumatism (EULAR) guidelines, IL-17-targeted therapy can be used in PsA patients with peripheral arthritis not/inadequately responding to at least one conventional synthetic disease-modifying antirheumatic drug and in whom anti-TNF therapy is not suitable for use. The Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) recommends for the use of any biologics (anti-TNF, anti-IL-12/23) in PsA patients with peripheral arthritis, even at an earlier stage if poor prognostic factors are present. IL-17-targeted therapy has been conditionally recommended, as Phase III data regarding secukinumab were not fully published at the time of publication of this guideline. For nail psoriasis, biologics (anti-TNF, anti-IL-12/23, or anti IL-17) are the preferred initial therapy. For the management of skin psoriasis, GRAPPA recommends biologics if conventional treatment fails; however, in severe disease, biologics can be used as the initial therapy. For dactylitis, biologics (anti-TNF, anti-IL-12/23) are recommended if conventional therapy fails or even as initial therapy. Anti-IL-17 is again conditionally recommended. For enthesitis, biologics or non-steroidal anti-inflammatory drugs can be used in non-responders to physiotherapy.
Conclusions
Currently available data are clearly indicative of the significant efficacy of anti-IL-17 monoclonal antibodies in reducing symptoms of PsA. Three-year data of secukinumab has also shown sustained and incremental efficacy for this agent. This drug has an acceptable safety profile and inhibitory effect on the radiographic progression in PsA. Secukinumab is approved by both the U.S. Food and Drug Administration (FDA) and EMA for the management of moderate to severe PsA. In their recent guidelines, both EULAR and the GRAPPA have included anti-IL-17 therapy for the management of PsA. On the contrary, no efficacy or safety data of secukinumab is available beyond 156 weeks. Data regarding the long-term effects on radiographic progression are also lacking beyond this duration, as well as regarding treatment duration and drug-free remission status. Results of ongoing trials may elucidate these lines of inquiry.