Abstract
This case details a 33-year-old female who presented with a suggestive autoimmune history, arthralgias, and splenomegaly, and tested positive for Sjögren’s syndrome and anti-liver–kidney microsomal antibody. This was further validated by findings from a liver biopsy, confirming a very rare association with Type 2 autoimmune hepatitis. Primary Sjögren’s syndrome is a sporadic disease with a global prevalence of 61 per 100,000 people and a total prevalence of 0.4% for secondary Sjögren’s syndrome. The prevalence of autoimmune hepatitis in association with primary Sjögren’s syndrome is 4–47%. It is divided into two types, associated with characteristic antibodies. Type 2 autoimmune hepatitis is rarely reported with Sjögren’s syndrome; much of the association reported in the literature has been with Type 1 autoimmune hepatitis.
INTRODUCTION
Autoimmune hepatitis is a condition closely related to autoimmune diseases. Previously, it was known to have three distinct types, which were recently further refined into two distinct types, each associated with characteristic antibodies. Sjögren’s syndrome is an autoimmune disease1 infrequently associated with autoimmune hepatitis; when it is, there is an association with the Type 1 subset of antibodies, such as antinuclear antibody (ANA), anti-smooth muscle antibody, perinuclear antineutrophil cytoplasmic antibodies, or antimitochondrial antibody, which usually is associated with primary biliary cirrhosis.2 Type 2 autoimmune hepatitis is associated with the anti-liver–kidney microsomal (LKM) antibody. This case presented with a suggestive history of Sjögren’s syndrome and additional workup revealed massive splenomegaly, which was further characterised as a feature of autoimmune hepatitis supported by the serological testing. An enlarged spleen could have multiple causes and implications to be ruled out in this present case, including myeloproliferative disorders, and Leishmaniasis, HIV, and Cytomegalovirus infections. The unique feature about this case was an association with Type 2 autoimmune hepatitis, which is overall less prevalent than Type 1 autoimmune hepatitis,3 and the association of Type 2 autoimmune hepatitis with Sjögren’s syndrome was a very rare entity in the literature review.
CASE PRESENTATION
A 33-year-old female of Asian ethnicity presented, without prior known comorbidities, with arthralgias, low-grade fever, stinging, foreign body sensation in eyes, and dry mouth for a long time. The patient denied photosensitivity, oral ulcers, alopecia, or history of abortions. Along with these symptoms, the patient had unintentional weight loss, anorexia, and abdominal discomfort preceded by abdominal fullness. She had no history of drug allergies or blood transfusion and was not taking any medications or alcohol. The examination was unremarkable, except for mild pallor along with hepatosplenomegaly. The patient was initially managed with low-dose intravenous antipyretics for the fever, oral nutritional supplements, and was given artificial tears for eye symptoms.
Laboratory workup showed a haemoglobin of 7.7mg/dL, mean corpuscular volume of 82 fL, total leukocyte count of 2.2 cells/μL with differentials of 61% neutrophils and 27% lymphocytes, and platelet count of 49 x109/μL of blood. It also showed increased erythrocyte sedimentation rate (83 mm/hour); elevated transaminases (normal range: <45 IU/L), aspartate aminotransferase: 213 IU/L; alanine aminotransferase: 239 IU/L; total bilirubin: 0.8 mg/dL; γ-glutamyltransferase: 20 IU/L; alkaline phosphatase: 165 IU/L; normal albumin: 3.7 g/dL, serum creatinine: 0.9 mg/dL; and prothrombin time: 12.8 sec. Hypergammaglobulinaemia was noted and there were elevated serum IgG levels of 2,712 mg/dL. Serology for viral infections, HBsAg, anti-hepatitis C (HCV), hepatitis B core antibody IgM, HIV, and Cytomegalovirus, was negative. Further workup showed positive ANA with titres of 2+ (granular), negative double-stranded-DNA, rheumatoid factor of 81 U/mL, strongly positive anti-Sjögren’s syndrome-A antibodies with titres of 41 U/mL, anti-Sjögren’s syndrome-B antibodies with titres of 14 U/mL, and anti-LKM was positive on two occasions with the presence of adequate negative and positive controls for immunofluorescence.
Ultrasonography of the abdomen was consistent with the above-mentioned examination findings; hence, CT of the abdomen was carried out to rule out intrahepatic vascular or thrombotic changes and was negative except for massive splenomegaly of 21.7 cm (Figure 1). The next aim was to obtain a liver biopsy, which demonstrated lymphoplasmacytic infiltrate with prominent plasma cells in the portal tracts with marked interface activity and multiple areas of hepatic necrosis consistent with autoimmune hepatitis (Figure 2). The sonographic findings of major salivary glands were remarkable for multiple hypoechoic lesions with high vascularity, thereby supporting the diagnosis of Sjögren’s syndrome in association with autoimmune hepatitis. Rose Bengal staining by ophthalmology remains the confirmatory diagnostic test of Sjögren’s; but, no objective measurements of oral and ocular dryness were performed. The biopsy of the labial glands was deferred by the patient.
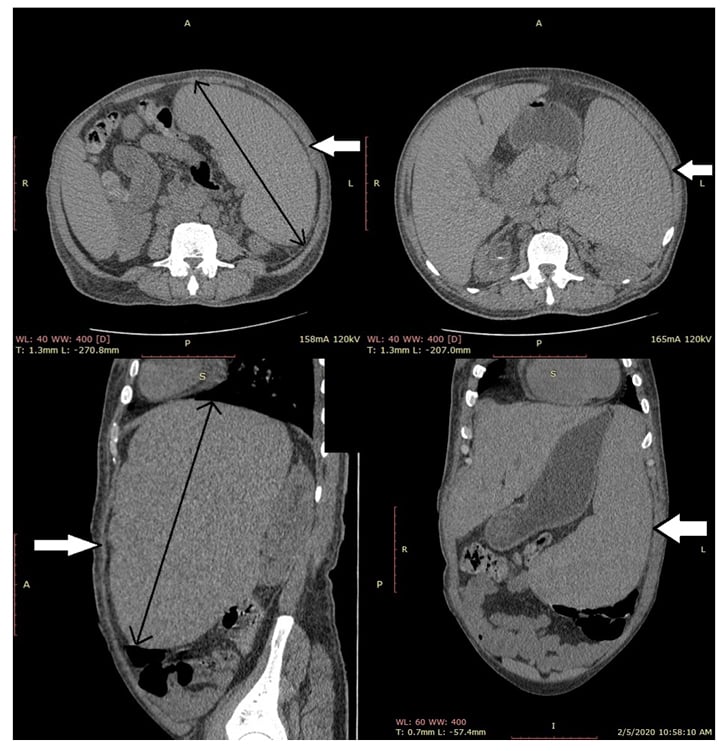
Figure 1: Axial, coronal, and sagittal sections of an abdominal CT scan showing splenomegaly.
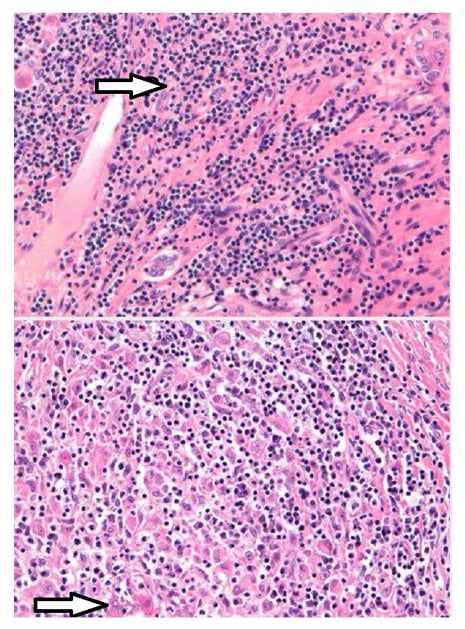
Figure 2: Histopathology showing lymphoplasmacytic infiltrate with marked interface hepatic necrosis.
The patient was started on prednisolone (60 mg/day), according to the patient’s weight, for 1 month and azathioprine (50 mg/day) after 2 weeks of treatment initiation. The response was monitored by improving cytopenias, the gradual return of liver enzymes to normal limits, and decreased IgG levels. After 2 months of treatment, steroids were tapered off on a follow-up visit and the patient was kept on azathioprine (100 mg/day) for a further 6 months. Laboratory workup at follow-up showed haemoglobin of 9.6 mg/dL, mean corpuscular volume of 84 fL, total leukocyte count of 4.5 cells/µL with differentials of 67% neutrophils and 24% lymphocytes, platelet count of 146 x 109 /µL of blood, aspartate aminotransferase 43 IU/L, and alanine aminotransferase 39 IU/L, with no fever documented.
DISCUSSION
Primary Sjögren’s syndrome is a sporadic disease with a global prevalence of 61 per 100,000 people with peak incidence in Europe and a total prevalence of 0.4% for primary and secondary Sjögren’s syndrome.1Sjögren’s syndrome is an autoimmune disease affecting many systems in the body, resulting in lymphocytic infiltration of salivary and lacrimal glands, progressing towards exocrine failure and fibrosis. Sjögren’s syndrome is categorised by the primary and secondary syndrome relating to autoimmune diseases including systemic lupus erythematosus, systemic sclerosis, rheumatoid arthritis, primary biliary cirrhosis, and autoimmune hepatitis.1-4 The most encountered clinical manifestations of Sjögren’s syndrome are keratoconjunctivitis sicca, xerostomia, salivary gland hypertrophy, rashes and skin oversensitivity, vaginal dryness with dyspareunia, arthralgias, Raynaud’s phenomenon, generalised osteoarthritis, and exhaustion. Less prevalent manifestations include low spiking fever, anaemia, leukopenia, vasculitis, thrombocytopenia, interstitial lung disease, cryoglobulinaemia, peripheral neuropathy, and glomerulonephritis.5 The predicted age of disease onset is 40–50 years, and most patients are females.1 Sjögren’s syndrome also targets nonexocrine organs such as the lungs, thyroid, kidney, liver, and pancreas, and the central nervous system.2,6,7
The American College of Rheumatology (ACR)-European League Against Rheumatism (EULAR) criteria are widely accepted for the diagnosis of Sjögren’s syndrome. They are defined as a score of ≥4 in relation to diagnostic features. This is along with labial salivary gland biopsy displaying lymphocytic infiltrates, positive anti-Sjögren’s-syndrome-related antigen, also known as anti-Ro, antibodies, an ocular staining score of >5 in at least one eye, positive Schirmer’s test with <5 mm in a span of 5 min, and salivary flow rate <0.1 mL/min with dry eyes and mouth. These criteria apply to any patient with at least one symptom of ocular or oral dryness. Conditions excluded from criteria are those with sarcoidosis, active HCV infection, amyloidosis, acquired immunodeficiency syndrome, IgG4-associated disease, graft-versus-host disease, and with radiotherapy of the head and neck.3,7,8 Treatment modalities for Sjögren’s syndrome include tear and saliva replacements, cevimeline, cyclosporine, pilocarpine, corticosteroid eye drops, and topical fluoride.6,9 Factors prognostic of distressing outcomes in Sjögren’s syndrome include declining levels of C4 complement, purpura, and mixed monoclonal cryoglobulinaemia.5 The hepatic manifestation of Sjögren’s syndrome includes primary biliary cirrhosis, HCV, nonalcoholic fatty liver disease, and autoimmune hepatitis.2,6,10
Autoimmune hepatitis is an unsettling and advancing disease of unexplained aetiology comprised of hypergammaglobulinaemia, autoantibodies, and hepatitis. It is characterised by the gradual disintegration of liver parenchyma.2,11,12 Autoimmune hepatitis is prevalent predominantly in females and often reactive to immunosuppressive modalities of treatment.2,3,12 It is associated with human leukocyte antigen (DR-3 and -4).11,13 The prevalence of autoimmune hepatitis in the USA is reported in approximately 100,000–200,000 inhabitants.12,14 The prevalence of autoimmune hepatitis in association with primary Sjögren’s syndrome is 4–47%.7 Autoimmune hepatitis is categorised into two types: Type 1 and Type 2 autoimmune hepatitis.11,15 Type 1 autoimmune hepatitis is generally characterised by the detection of ANA, smooth muscle autoantibodies (SMA), and perinuclear-antineutrophil cytoplasmic antibodies (p-ANCA).16,17 Type 2 autoimmune hepatitis is characterised by the detection of definitive antibodies such as anti-LKM Type 1; detection of anti-LKM antibody Type 3 or antibodies against liver cytosol specific Type 1 antigen are infrequently found.17 Type 1 autoimmune hepatitis is the classic form of the disease that usually responds well to corticosteroids. Type 2 autoimmune hepatitis is typically seen in females, the paediatric, and Mediterranean population, and can present as severe disease, known as fulminant hepatitis. The prevalence of ANA in patients with Type 1 autoimmune hepatitis associated with Sjögren’s syndrome according to Fayaz et al.18 is 1.70–13.00%, while 1.49% of patients are found to have SMA. Tzioufas et al.17 quoted the prevalence of ANA in patients with Type 1 autoimmune hepatitis associated with Sjögren’s syndrome as 77.0–90.0%, while 6.5–62.0% of patients were detected with anti-smooth muscle antibody.17,18 Gatselis et al.11 quoted the prevalence of anti-LKM Type 1 antibody in patients with Type 2 autoimmune hepatitis associated with Sjögren’s syndrome as 0–10%, while the prevalence of anti-LKM Type 3 antibody as 5–10%.11 Zeron et al.19 quoted 0 prevalence of autoimmune of anti-LKM Type 1 antibody type 1 as none of the patients had been detected with this autoantibody.19
Clinical manifestations of autoimmune hepatitis vary with the severity of disease, ranging from no visible symptoms of liver disease to severe or acute fulminant hepatitis. The foremost assessment is either normal, or symptoms of chronic liver disease present in the form of hepatomegaly, splenomegaly, palmar erythema, and spider naevi. When the disease precipitates it can lead to the development of ascites, oesophageal varices, portal gastropathy, and cytopenias.20,21 These features were also present in this case, with portal gastropathy causing massive splenomegaly and ultimately cytopenias. Autoimmune hepatitis shows a bimodal age of outbreak with children and adolescents at one age, and middle-aged females at the fourth–sixth decade of life, especially after menopause.22 Diagnostic criteria cited for autoimmune hepatitis is a scoring system devleoped by the International Autoimmune Hepatitis (AIH) Group and the International Association for the Study of the Liver (IASL).12 Laboratory investigations of Sjögren’s syndrome comprise biochemical and immunological tests, including levels of liver enzymes along with plasma levels of IgM, IgG, and IgA. Immunological tests, including the detection of ANA, anti-smooth muscle antibody, and anti-LKM, are done by direct immunofluorescence assay. Percutaneous liver biopsy is also performed.7,10 Histological findings of autoimmune hepatitis show interface hepatitis, lymphoplasmacytic infiltrate, and rosette formation of liver cells, plasma cells, and piecemeal necrosis.2,12 Treatment options to cure autoimmune hepatitis include monotherapy or a combination of corticosteroids, such as prednisolone, along with immune-modulating agents, such as azathioprine.12
Complications of autoimmune hepatitis include rheumatoid arthritis, Hashimoto’s thyroiditis, and Sjögren’s syndrome, along with unusual complications of antiphospholipid antibody syndrome.23 In this present case, there were vague sicca symptoms in conjunction with a moderate level of ANA and anti-Sjögren’s syndrome-A antibody, a low titre anti-Sjögren’s syndrome-B antibody, and an ultrasound suggestive of Sjögren’s. Most patients with Sjögren’s disease will have a strongly positive ANA and anti-Sjögren’s syndrome-A antibody at very high titres; hence, the symptoms described may be of secondary Sjögren’s disease in association with autoimmune hepatitis. There was no evidence of renal involvement with normal creatinine levels; however, systemic lupus erythematosus was in the differential diagnosis and might be the cause of secondary Sjögren’s disease, but the antibody panel showed negative double-stranded-DNA.
CONCLUSION
The overall frequency of Type 2 autoimmune hepatitis is less than that of Type 1 autoimmune hepatitis, but it can be associated with autoimmune diseases such as Sjögren’s syndrome. Although the majority of the association between autoimmune hepatitis and Sjögren’s syndrome reported in the literature has been with Type 1 autoimmune hepatitis, this case presented with positive anti-LKM antibodies and with Sjögren’s syndrome, making it a very rare and novel association with Type 2 autoimmune hepatitis.