Abstract
The phenotypical overlap between the spondyloarthropathies (SpA) and Crohn’s disease (CD) has long been recognised. More recently, the co-inheritance of these diseases and the existence of a plethora of shared genetic risk loci have been demonstrated by genealogic databases and genome-wide association studies. Now there is mounting evidence to suggest that the interplay between the gut microbiota and host genetics is central to the shared aetiopathogenesis of SpA and CD. The clinical management of patients with both SpA and CD can be challenging. Preliminary studies seeking to understand this interplay have identified novel therapeutic targets and approaches, which may, in the future, significantly advance patient care. This review provides an overview of the role of host genetics and the intestinal microbiota in the shared aetiopathogenesis of SpA and CD, and explores how this interplay can advance the search for new therapeutic targets.
INTRODUCTION
The spondyloarthropathies (SpA) are a spectrum of inter-related conditions that include axial spondyloarthritis (AxSpA), (also known as ankylosing spondylitis; AS), non-radiographic axial spondyloarthritis, peripheral spondyloathritis (pSpA), reactive arthritis (ReA), and enteropathic arthritis associated with Crohn’s disease (CD).1,2 Phenotypically, AS and CD share many clinical features. Extra-intestinal manifestations of CD include both a peripheral arthritis and a sacroiliitis, which is a hallmark of SpA, whilst AxSpA and pSpA are associated with evidence of microscopic intestinal inflammation on biopsy and capsule endoscopy;2 one study demonstrating this to be present in 42% of patients with SpA.3,4 Although this bowel inflammation is considered asymptomatic, it has been shown to strongly correlate with axial disease activity in AxSpA.3,5,6 Clinical inflammatory bowel disease (IBD) is also common in AxSpA, with a prevalence ranging from 6% to 14% in populations of European decent.6 The DESIR cohort noted a frequency of IBD of 2.6% in patients with early AxSpA.7 Interestingly, in Han Chinese this association is rare at 0.4%, suggesting a genetic and/or environmental difference between these populations.8 Similarly, SpA features are the most common extra-intestinal manifestations of CD, with approximately 10% of patients with CD developing AxSpA, 13% oligo-articular or poly-articular pSpA, and 5% other typical SpA features such as enthesitis and dactylitis.9
The development of classification criteria has enabled better characterisation of these conditions and thus the ability to study overlapping aetiological and therapeutic pathways.10 However, AxSpA in particular is often a challenging diagnosis to make, with a mean delay in diagnosis from onset of symptoms of 8–10 years.11
This review has two main aims: 1) to evaluate the evolving evidence for a shared aetiopathogenesis and, in particular, whether the interplay of host genes and the intestinal microbiota are central to this shared aetiopathogenesis; and 2) to discuss the importance of understanding this interplay in identifying potential new therapeutic targets.
THE SHARED GENETIC ARCHITECTURRE OF SPONDYLOARTHROPATHIES AND CROHN’S DISEASE
The seminal study of Thjodleifsson et al.12 was the first to provide evidence of the existence of a common genetic background between SpA and CD. In this study of the Icelandic Genealogy Database, first- and second-degree relatives of patients with AS had a 3.1 and 2.0 increased risk of developing CD, respectively, compared to the general population.12 Near-identical cross-risk ratios for developing AS were found for first- and second-degree relatives of patients with CD.12
The estimated heritability of AS by twin studies is >90%,13 with the Class I major histocompatibility complex (MHC) molecule human leukocyte antigen (HLA)-B27 accounting for much of this risk and representing one of the strongest genetic associations with any common human disease.14 The prevalence of HLA-B27 is 80–95% in patients of European ancestry with AS, compared with a general population prevalence of 6–16%14-17 in central and Northern European countries. Interestingly, whilst HLA-B27 is not considered a risk gene for CD, 25–78% of patients with CD who carry this gene will go onto develop AS and another 7–15% will develop an isolated sacroiliitis.6
The era of genome-wide association studies (GWAS) has significantly advanced the search for a shared genetic architecture between SpA and CD by identifying a plethora of additional risk loci for these diseases. The risk loci discovered by GWAS have underscored the importance of bacterial detection and handling in the development of both SpA and CD.
An early, watershed study in characterising this shared genetic architecture was Danoy et al.18 Taking the 53 genetic loci most strongly associated with CD in three separate GWAS, Danoy et al.18 demonstrated that eight were also significant risk loci for AS, namely IL23R (rs11465804), IL12B (rs10045431), CDKAL1 (rs6908425), LRRK2 (rs11175593), chr13q14 (rs3764147), chr1q32 (rs11584383), and STAT3 (rs6503695, rs744166) (Table 1).19,20 This constellation of genetic variants highlights the potential importance of the pro-inflammatory IL-23/IL-17 pathway and intestinal microbiota in the shared aetiopathogenesis of CD and SpA. IL-23 expression has been shown to be upregulated in the presence of gut dysbiosis in both the terminal ileum of patients with SpA and patients with CD.21 Elevated circulating levels of IL-23 lead to the activation of STAT3, which induces the expression of IL23R on the surface of Th17 cells.22 This receptor enables these pro-inflammatory cells to secrete IL-17 in response to IL-23.22 Significantly higher levels of IL-17 have been noted in the intestinal mucosa and joints of both patients with SpA and patients with CD compared to healthy controls.21 Furthermore, in patients with SpA, IL-17 levels have been shown to directly correlate with disease activity (Table 1).23 In a similar fashion, functional studies have demonstrated that LRRK2 plays an important role maintaining gut and joint homeostasis through the clearance of gut pathogens, such as Salmonella typhimurium. This bacterium has been demonstrated to trigger reactive arthritis by causing the activation of the NLRC4 inflammasome in macrophages (Table 1).24
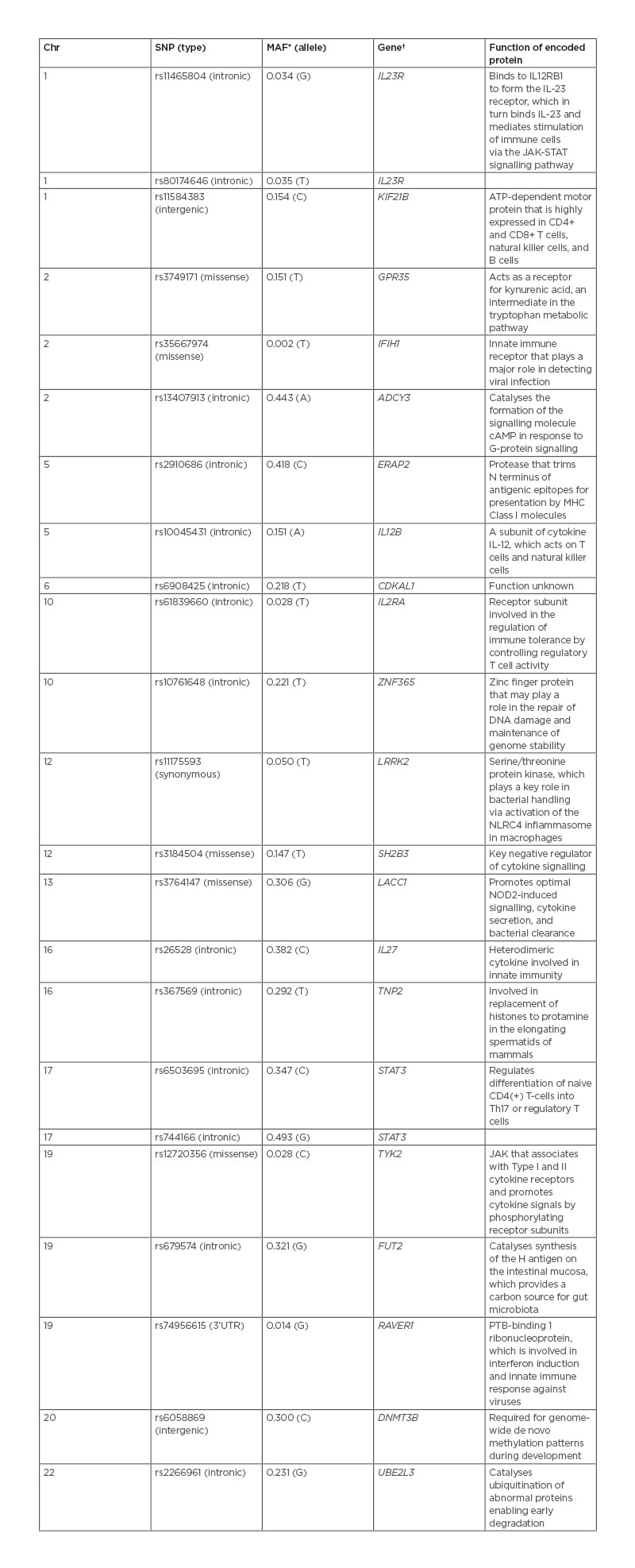
Table 1: Genetic susceptibility loci strongly associated with both ankylosing spondylitis and Crohn’s disease.
*Minor allele frequency in the ‘1,000 Genome’ Phase III Combined Population.20
†Gene candidate nearest to the index SNP.
cAMP: cyclic adenosine monophosphate; Chr: chromosome; MHC: major histocompatibility complex; SNP: single-nucleotide polymorphism; STAT: signal transducer and activator of transcription proteins.
The association of 1q32 single-nucleotide polymorphism (SNP) rs11584383 with CD and SpA has been independently replicated,25 but its significance remains to be established. This intergenic SNP is postulated to be in linkage disequilibrium with a functional SNP in a nearby gene.25 The most likely candidate is Kinesin Family Member 21B (KIF21B). This gene is highly expressed in CD4+ and CD8+ T cells, B cells, and natural killer cells.26 SNPs and copy number variants within KIF21B have subsequently been strongly associated with AS susceptibility in Korean and Han Chinese patient cohorts.19,27
The delineation of a shared genetic architecture has been further advanced by Ellinghaus et al.28 Utilising high-density immuno-chip genotyping data from five spondyloarthritis spectrum and associated diseases (AS, CD, ulcerative colitis [UC], psoriasis, and primary sclerosing cholangitis), this study identified a total of 187 non-MHC risk loci, which were shared between two or more of these diseases.28 Among the strongest shared AS/CD loci were the intronic SNP rs2910686 in ERAP2, the missense SNP rs12720356 in TYK2, and the intronic variant rs679574 in FUT2 (Table 1). These associations again point to the importance of the gut microbiome and mucosal immunity in both AS and CD. Endoplasmic reticulum aminopeptidases 2 (ERAP2), along with ERAP1, are key players in the adaptive immune response of the gut and the joint.29 These enzymes trim peptides arising from infection or cell damage. The ERAP1- and ERAP2-trimmed peptides are then loaded onto MHC-1 molecules and displayed on the surface of affected cells, triggering a T-cell-mediated immune response against these cells in the gut mucosa or joint.29 Pepelyayeva et al.,30 using an ERAP1 knockout mouse model, demonstrated that ERAP proteins play a central role in both preventing inflammation and in maintaining homeostasis of gut microbiota. ERAP1-deficient mice were significantly more likely to develop severe colitis after dextran sulphate sodium challenge compared to wild-type mice. Furthermore, this knockout model exhibited marked gut dysbiosis and developed many of the hallmarks of axial SpA including sacroiliitis, joint erosions, and spinal ankylosis.29,30 Tyrosine kinase 2 (TYK2) is an intracellular signalling protein, which belongs to the JAK family.31 This protein plays a pivotal role in the expansion and maturation of Th17 cells through the activation of IL23R–STAT3 signalling.31 Murine models of colitis and SpA have demonstrated the specific inhibition of TYK2 ameliorates both intestinal and joint inflammation.31 Fucosyltransferase 2 (FUT2) is responsible for the synthesis of the H antigen on the intestinal mucosa; this H antigen is an oligosaccharide moiety that acts as both an attachment site and carbon source for the intestinal microbiome.32 Individuals who are homozygous for loss-of-function alleles of FUT2 exhibit significant intestinal dysbiosis and have increased susceptibility to CD.32
Collectively, the findings of these genetic studies suggest CD and AS do indeed have significant shared genetic architecture (Table 1), and that shared clinical characteristics relate to shared pathophysiological pathways. These studies also highlight the possibility that concomitant disease may represent a genetically distinct population.
GENE MICROBIAL INTERACTIONS IN ANIMAL MODELS AND HUMANS
Several, not necessarily mutually exclusive, theories have been proposed to explain the overlap between AS and CD, including the gut–joint axis of inflammation (Figure 1).33 Central to this hypothesis is the concept that mucosal immunity and the perturbations in the intestinal microbiome in genetically susceptible individuals may instigate and perpetuate both gut and joint inflammation.33
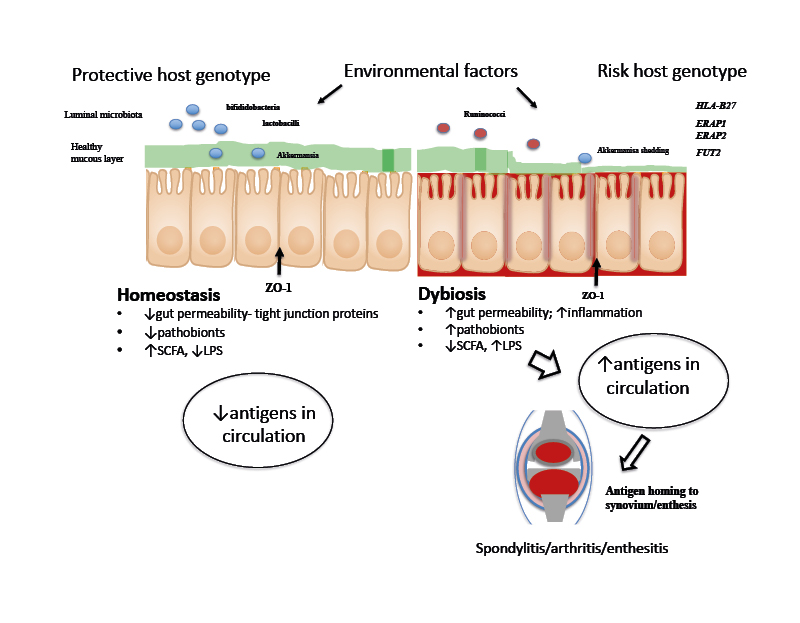
Figure 1: A proposed schema for the role of gut microbiota and host genetics in gut and joint inflammation.
Gut inflammation and increased epithelial permeability result in dysbiosis, mucin layer degradation, and tight-junction protein abnormalities (e.g., ZO-1). Transposition of luminal antigens into the lamina propria stimulates pro-inflammatory pathways and antigens are presented by dendritic cells through HLA Class I proteins. High antigenic loads lead to translocation of these antigens to the joint, resulting in aberrant inflammation.
HLA: human leukocyte antigen; LPS: lipopolysaccharide; SCFA: short chain fatty acids; ZO-1: zonula occludens 1.
The first evidence to support this hypothesis came from studies in HLA-B27 transgenic rats.34 These rats developed a spondylitis-like syndrome under normal conditions; however, when raised in a germ-free environment, colitis, arthritis, and skin disease associated with this animal model dramatically improved.34 Certain bacterial strains, when introduced to these germ-free rats, were found to maintain remission while other bacterial strains caused relapse.35 A more recent SpA–CD animal model, the SKG mouse, provided further evidence of the role of host genetics and microbes in the development of SpA and IBD. This model is a BALB/c mouse strain that carries the SKG ZAP-70 W163C mutation.
Under germ-free conditions, SKG mice remain healthy; however, when exposed to curdlan, an antigen derived from the cell walls of yeast, fungi, and bacteria, the W163C mutation causes SKG mice to develop a SpA-like disease with arthritis and small intestinal inflammation.36 The SKG mouse model was developed to study auto-reactive CD4+ T cells via an IL-23-dependent pathway known to be pivotal in the pathogenesis of both IBD and SpA.37
Observations made in ReA provide further evidence to support a central role for host genetics and microbes in both SpA and IBD, and the existence of the gut–joint axis of inflammation. ReA, part of the SpA spectrum, can be triggered by an enteric infection with a number of organisms including Salmonella and Shigella.38 Individuals who are HLA-B27-positive are more than five times more likely to develop an arthritis after exposure to a triggering organism than those who do not carry this allele.39 The exact mechanism for this is still speculative, but Salmonella antigens have been shown to be immunogenic to mononuclear cells within joint synovial fluid.40
INTESTINAL MICROBIOTA AND ITS ROLE IN DISEASE IN PATIENTS WITH SPONDYLOARTHROPATHIES
The human intestine contains a unique microbiota, which maintains a relatively stable ecological state with a dynamic equilibrium. An individuals’ microbiota is highly resistant to change, with key species maintained over long periods. However, microbial profile and abundances do change over a persons’ lifetime, with diet and other exogenous factors influencing its composition. The overall resilience of the resident flora can be impacted by major perturbations, which can lead to a ‘tipping point’ beyond which homeostasis fails and significant shifts in microbial profiles can occur.41 The colonising microbiota is largely acquired at birth, although some elements may even be acquired in utero.42 Mode of delivery, whether vaginal or via Caesarean section, can influence early acquisition of the microbiota.43 This microbiota is essential to the development of the intestinal immune system, which in turn influences the constituents of the microbiota.44 A recent comprehensive study of the human gut microbiota identified 204,938 non-redundant genomes from 4,644 gut microbes, many of which are specific to individual human populations.45 Moreover, metabolomics studies have demonstrated that the small-molecule metabolites produced by the microbiota can profoundly alter host–gut microbiota interactions and thereby intestinal inflammation, and potentially joint inflammation (Figure 1).44
Transient or permanent perturbations, or dysbiosis, of the gut microbiota have been associated with CD, UC, and irritable bowel syndrome. Attempts have been made to quantify these perturbations, such as the GA-map™ Dysbiosis Index (Biohit Healthcare Ltd, Helsinki, Finland), which uses faecal samples from patients and compares the result with population norms,46 although such approaches have been criticised for not recognising the dynamic nature of the gut microbiota.47
An important, unresolved question is whether the gut microbiota is a causative factor for gut inflammation in SpA or whether changes in the microbiota are a product of inflammation. Animal models seem to suggest a causative role for the interplay of gut microbiota with host genetics. However, host genetics appear to play only a minor role in determining the composition of the microbiota, as shown by twin studies, accounting for no more than 8% of microbiota variability. Furthermore, unrelated individuals in the same household share common constituents, with approximately 20% of the variability in the microbiome between individuals resulting from diet, drug therapies, and body habitus.48 These findings raise the question of whether it is possible to manipulate the gut microbiota before the development of clinical disease.
Several studies have investigated the gut microbiota in SpA. One study looked at the dysbiosis of faecal microbiota in 150 patients with AS, 18 patients with UC, and 17 healthy controls.49 Dysbiosis was defined as a Dysbiosis Index of ≥3. Using this definition, 87% of patients with AS had dysbiosis.49
A recent study used shotgun meta-genomic sequencing and metabolomics to analyse faecal microbiota in a case-control cohort of 250 subjects.50 Cases and controls had different microbiota profiles, with a prevalence in AS patients of bacterial species thought to be pro-inflammatory.50 Furthermore, the gut microbiota of patients with AS carried a higher load of bacterial peptides known to be presented by HLA-B27, suggesting either HLA-B27 fails to clear these or that these peptides drive the immune response associated with HLA-B27.50
POTENTIAL NEW THERAPEUTIC TARGETS AND APPROACHES
Whilst most studies still focus on quantity and diversity of the microbiota, investigating the metabolic effects, or metabolomics, of the gut microbiota is increasingly recognised as having greater potential for developing new therapies.
Mucin Barrier
An example of this more targeted approach to microbiota analysis is illustrated by studies of the mucin barrier in the gut. In the healthy gut, bacteria predominantly reside in the gut lumen, with few able to penetrate into the mucus layer. Mucin 2 (Muc2) is the main constituent of the mucus layer, the integrity of which is maintained by glycosylation reactions within the intestinal epithelial cells, which are stimulated by IL-22. This process seems to be key in maintaining the equilibrium between host and gut bacteria (Figure 1). The microbiota induces expression of host fucosyltransferase 2 and a deficiency of fucosyltransferase 2 has been noted to cause migration of leucocytes into the caecum epithelium following infection with S. typhimurium.51
The importance of an intact mucus layer for gut microbial tolerance is demonstrated by numerous studies. Attenuation of the mucin barrier is a stimulus for intestinal inflammation. A thin mucus layer is associated with increased bacterial invasion in IBD. Mouse models deficient in Muc2 induce the Th17 cells and Th1 responses, and reduced Muc2 levels are detectable in patients with UC prior to the onset of inflammation.51
The mucin-degrading commensal Akkermansia muciniphila is of particular interest. This bacterium resides in the mucus layer and can restore mucus thickness and the mucin barrier; it is also thought to contribute to improving the symbiosis between host and microbiota.52 A reduction in the abundance of A. muciniphila and a corresponding increase in Ruminococcus species is seen in CD. R. gnavus produces an inflammatory polysaccharide, produced in abundance during flares of CD.53 An increase in Ruminococcus species is also seen in SpA.54
Fucose, a natural monosaccharide, has been shown to ameliorate colitis in an experimental animal model by improving dysbiosis, including a reduction of Ruminococcus species.55 There is also interest in using A. muciniphila as a probiotic species (see later).
Short Chain Fatty Acids
Dietary fibres are metabolised by colonic bacteria to short chain fatty acids (SCFA), such as lactate. These metabolites are essential for maintaining immune tolerance and promoting symbiosis between the host and the gut microbiota. SCFAs inhibit intestinal inflammation by mediating the host immune response and may also prevent pathogen colonisation.51 Reduced levels of SCFA have been found in the faecal samples of patients with IBD.56 Furthermore, in the HLA-B27 transgenic rat model the intestinal metabolome differs from wild-type rats within weeks of birth. In this model SCFAs attenuate inflammatory disease.57
Lipopolysaccharides
Lipopolysaccharide (LPS) is expressed on the outer surface of Gram-negative bacteria and is a crucial signalling molecule recognised by immune cells and binds to CD14 and TLR4 receptors on these cells. Modification of LPS enables pathogens to evade the immune system. LPS also directly induces intestinal inflammation, disrupting intestinal tight junctions and increasing gut permeability. Dysbiosis has been shown to increase the prevalence of pathobionts and LPS in patients with IBD.51 In AS, the increase in gut permeability is associated with the translocation of LPS and other bacterial products across the mucosa, with LPS stimulating IL-23 production.58
APPROACHES TO THE CLINICAL ASSESSMENT AND MANAGEMENT OF BOWEL INFLAMMATION IN PATIENTS WITH SPONDYLOARTHROPATHIES
There is increasing evidence to suggest that patients with SpA and concomitant bowel inflammation may be a genetically and microbiome-distinct patient subgroup.33 This subgroup presents a number of significant clinical challenges. Identifying patients with AS/SpA who have bowel inflammation is challenging.5 Recommendations to improve identification of patients with coexisting IBD/SpA include education of primary care physicians and developing shared clinics with gastroenterologists.11 Some centres have trialled clinical pathways in an attempt to more readily identify these patients.59 A combination of patient questionnaires (such as the Dudley Inflammatory Symptoms Questionnaire [DISQ]), faecal calprotectin, and CRP may help to identify patients who should go forward for more invasive studies such as colonoscopy or capsule endoscopy.5,59 A further opportunity to identify patients with sacroiliitis is afforded by the routine assessment of the sacroiliac joints in patients with IBD who undergo MR enterography to evaluate the extent and complications of their IBD. Small studies have shown this is feasible and an effective way of identifying patients with sacroiliitis and further studies are planned.60
Concomitant IBD and SpA has implications for management using existing standard therapies. Non-steroidal anti-inflammatory drugs may worsen bowel inflammation. Similarly, the TNF receptor blocker etanercept and the IL-17 monoclonal antibody secukinumab, which are effective for treating moderate to severe AS, do not ameliorate, and in the latter case may actually worsen, bowel inflammation. To date, the established monoclonal antibody therapies such as adalimumab and infliximab are the best therapies for IBD/SpA overlap.61 More recently, evidence for favourable outcomes in both AS and IBD has also been seen in studies of JAK inhibitors such as tofacitinib.62
MICROBIOTA RESEARCH REVEALS POTENTIAL FUTURE THERAPIES
At present, the state of knowledge is insufficient to recommend specific treatment modalities in AS/SpA. However, existing therapies have been shown to affect the microbiota and several areas of research show promise for future therapies.
Prebiotics
Prebiotics are substrates not digested in the human small bowel but available to the microbiota. Supplementary dietary fibre and anaerobic fermentation produces SCFA, which may promote a selective pressure towards a beneficial colonic microbiota. Prebiotics have shown benefit in several animal models of gut inflammation.56 No studies are available in AS, but several small studies have been undertaken in IBD. In one of these, a double-blind pilot study in patients with active UC, using oral inulin, an oligofructose supplement, a decrease in symptoms and reduced faecal calprotectin levels were noted.56
Exclusive Enteral Nutrition
Exclusive enteral nutrition (EEN) has been used widely in paediatric CD. It has shown particular promise in recent-onset CD, resulting in higher remission rates and a trend towards better growth, although relapses and complication rates were similar to patient cohorts receiving conventional therapy.64 However, a Cochrane review of adults with established quiescent CD could make no firm conclusions regarding the efficacy or safety of EEN.1
A more recent study of diet intervention, which replicated elements of EEN, showed changes in the microbiota and a decrease in gut inflammation in patients with active CD.65 To date, there have been no studies of EEN in SpA.
Probiotics
A single, small, randomised trial in AxSpA of an oral probiotic containing Bifidobacteria, a lactobacillus, and Streptococcus salivarius found no significant benefit in primary outcomes.66 However, since this study more promising probiotic candidates, specifically A. muciniphila have emerged. Currently studies exploring A. muciniphila as a therapeutic candidate have only been performed in animal models, but future studies in human IBD and SpA are likely.52
Faecal Microbiota Transplant
Faecal microbiota transplant involves the transfer of pre-screened minimally manipulated stool from healthy donors into the gastrointestinal tract of a recipient, via enema or nasogastric tube, with the purpose ameliorating dysbiosis. The place of faecal microbiota transplant in the management of colitis due to Clostridium difficile is well established and guidelines have been developed internationally for its use in this situation.67 Several small studies have shown promise in ameliorating colitis in UC, but as yet this has not been translated to therapy beyond clinical trials.68 A small study is underway in patients with psoriatic arthritis.69
CLINICAL AND RESEARCH CONSEQUENCES OF THE INTERPLAY OF GUT MICROBIOTA AND HOST GENETICS
Research priorities for the future should build on the expanding knowledge of the gut microbiota and gut inflammation. Improved risk stratification of close relatives of index cases, through polygenic risk scores, could identify individuals at high risk. Early assessment and microbial analysis may then identify individuals with modifiable dysbiosis. These individuals may be ideal candidates for novel microbial therapies designed to protect and enhance the mucin layer and select out pro-inflammatory microbiota. There is much work to be done in human populations to delineate the interplay between host genetics and the microbiota, but preliminary results show definite promise for an effective preventive approach in the future.
KEY LEARNING POINTS
- Significant phenotypic overlap exists between SpA and CD.
- Patients with SpA and concurrent CD present clinical management challenges.
- There is increasing evidence to suggest that the interplay between host genetics and gut microbiota is central to this overlap.
- Understanding this interplay may provide novel therapies for improved management of concurrent SpA and CD.