Abstract
Type I interferons (IFN) are widely expressed cytokines that play a pivotal role in the cell-intrinsic antimicrobial process, especially in viral infections. Studies have shown an increased expression of Type I IFNs and their induced genes in peripheral blood cells and exocrine glands from patients with Sjögren’s syndrome (SS), indicating that the Type I IFN pathway a vital role in the pathogenesis of this disease.
The source of upregulated Type I IFNs in patients with SS is unknown. Many cells were reported to contribute to the process, especially plasmacytoid dendritic cells and other innate immune cells.
The activation of Type I IFN signalling was regulated by both genetic and epigenetic pathways, suggesting that genetic predisposition and environmental factors may affect the initiation and progression of SS. Treatments targeting the Type I IFN pathway are still under evaluation and more results are needed to see their value.
The authors’ review aims to summarise the functions and regulations of Type I IFNs in the pathogenesis of SS. They also summarise current treatments (including clinical trials) targeting the Type I IFN pathway in treating SS and provide potential targets for future studies.
Key Points
1. Type 1 interferons (IFN) and their induced genes have been found to have increased expression in Sjögren’s syndrome (SS), with increased expression correlating with clinical features in SS.2. Activation of signalling for Type 1 IFNs and their induced genes is regulated by both genetic and epigenetic pathways; this suggests that genetic predisposition and environmental factors affecting IFN may alter onset and progression of SS.
3. Treatments that target blocking of Type 1 IFNs could provide a promising avenue for treatment of SS; results of ongoing clinical trials for SS and other auto-immune diseases are eagerly awaited.
INTRODUCTION
Interferons (IFN) are a class of cytokines that are produced in response to pathogenic stimuli.1 The IFN family was divided into three classes: Type I, Type II, and Type III. Type I IFNs are widely expressed cytokines that play a pivotal role in the cell-intrinsic antimicrobial process, especially in the context of viral infections.2 They mobilise innate immune responses by enhancing antigen presentation and cytotoxicity effect, while restraining the magnitude of inflammation to avoid toxicity.2 They can also affect the adaptive immune system by promoting the functions of T and B cells in various ways.2,3
In humans, IFN-Is consist of 13 IFN-α subtypes, IFN-β, IFN-κ, IFN-ε, IFN-δ, and IFN-ω. IFN-α is produced by haematopoietic cells, in particular plasmacytoid dendritic cells (pDC), while IFN-β can be produced by most cell types. Although protective in acute viral infections, IFN-Is, especially IFN-α and IFN-β, have been implicated as deleterious in autoimmune diseases.4,5
Meanwhile, Type II IFNs, also known as IFN-γ, are released by cytotoxic T cells and T helper cells, playing a major role in building cellular immunity and priming the other two types of IFNs.6
Type III IFNs include four IFN-λ subtypes, which are mainly expressed by mucosal epithelial cells. They activate the same receptor-associated JAK and similar downstream pathways as IFN-Is by binding to different receptors;7 thus, they may play a similar role as IFN-Is such as antiviral activity.8 These two types of IFNs are beyond the scope of this review.
Sjögren’s syndrome (SS) is a chronic autoimmune inflammatory disorder characterised by dryness of the eyes and mouth due to a functional impairment of the salivary and lacrimal glands. It has been found that lymphocytic infiltration and deposition of autoantibodies of exocrine glands cause chronic inflammation and functional impairment.9 SS may also affect multiple organs, and not just exocrine glands, such as the skin, joints, lungs, gastrointestinal tract, kidney, vessel, and haematology and nervous systems.10 Besides, chronic B cell stimulation in SS increases the risk of lymphoid malignancies,11,12 SS can occur as a lone condition (primary SS) or accompanied by another connective tissue disease (secondary SS); both have similar presentations.13 The pathogenesis of SS has yet to be fully elucidated.
In the past years, a number of studies have revealed the important role of IFNs in the initiation and progression of SS. Although the three types of IFNs have been implicated in the pathogenesis of SS, IFN-Is and their induced genes were correlated with clinical presentation, disease activity, and antibody titres of SS, enhancing our understanding of mechanisms of SS. This review focuses on the role of Type I IFNs, mainly IFN-α and IFN-β, and summarises their roles in the pathogenesis and treatment of SS.
TYPE I INTERFERONS SIGNALLING PATHWAY
Type I IFNs can be produced when stimuli like microbial products are sensed by various cellular receptors, which then induce IFN-stimulated gene (ISG) expression.2 Production of Type I IFNs depends on the cell types and environment.5 Innate immune cells such as macrophages and dendritic cells (DC) produce Type I IFNs after sensing stimuli by using varieties of pattern-recognition receptors (PRR), which function as sensors for pathogen-associated molecular patterns. Toll-like receptors (TLR) are one of the important types of PRR. They are transmembrane proteins and detect pathogen-associated molecular patterns derived from extracellular bacteria or bacteria that have been taken into vesicular. Other pathogens sensing systems include retinoic acid inducible gene I (RIG-1), melanoma differentiation-associated protein 5 (MDA5, which is also known as IFIH1), stimulator of IFN genes protein, and nucleotide-binding oligomerisation domain-like receptors (NLR).
It is widely acknowledged that pDCs are the major producers of IFN-α (Figure 1). In pDCs, single strand RNA activates endosomal TLR7 or TLR8, which then activates IFN regulatory factor (IRF) 7 and/or IRF5. Double strand DNA activates endosomal TLR9, or RNAs activate cytosolic nucleic acid sensors MDA5 or RIG-1, all of which result in activation of IRF7. Translocation of IRF5 to the nucleus induces transcription of Type I IFNs and pro-inflammatory cytokines such as IL-6 and TNF. Translocation of IRF7 induces transcription of Type I IFNs, especially a high amount of IFN-α. Type I IFNs then bind to the Type I IFN receptor (IFNAR) in pDCs and activate the canonical JAK signal transducer and activator of transcription (STAT) pathway, resulting in transcription of ISGs. ISGs include IRF7, which provides a feed-forward mechanism for the production of more Type I IFNs.

Figure 1: Major treatment targets of Type I interferon production in Sjögren’s syndrome.
In Sjögren’s syndrome, exocrine epithelial cells and infiltrating immune cells such as pDCs produce large amounts of Type I IFNs, inducing transcription of ISGs and pro-inflammatory cytokines through the JAK–STAT pathway. The overexpression of Type I IFNs induce B cell expansion and differentiation into plasma cells, which produce autoantibodies and cause organ damage. Type I IFN antibodies and IFN-α vaccines, which induce the production of IFN-α antibodies, inhibit the function of Type I IFNs. HCQ can prevent the activation of TLR7 and TLR9. TBK1i can prevent the activation IRF3 and IRF7. RNase digests circulating RNAs and inhibits Type 1 IFN production. JAKi and TYK2i are important in the downstream pathway of Type 1 IFNs and inhibit ISG expression.
BAFF B: cell activating factor; HCQ: hydroxychloroquine; IFNAR: Type I interferon receptor; IRAK: IL-1 receptor-associated kinase; IRF: interferon regulatory factor; ISG: interferon-stimulated genes; JAKi: JAK inhibitors; MAVS: mitochondrial antiviral signalling protein; MDA5: melanoma differentiation-associated protein 5; Myd88: myeloid differentiation factor 88; NAP1: nucleosome assembly protein 1; pDC: plasmacytoid dendritic cells; RIG-1: retinoic acid inducible gene 1; RNase: ribonuclease; SINTBAD: similar to NAP1 TBK1 adaptor; STAT: signal transducer and activator of transcription; TANK: TRAF family member associated NF-κB activator; TBK1i: TANK-binding kinase 1 inhibitor; TRF3: TATA-box-binding protein-related factor 3; TLR: toll-like receptor; TYK2: tyrosine kinase 2; TYK2i: tyrosine kinase 2 inhibitor.
In other innate immune cells such as phagocytes and DCs, stimulations come from both extra- or in-cell. Lipopolysaccharide binds to TLR4 on cytomembrane or double-stranded RNA, which activates endosomal TLR3 and will then activate IRF3 via TIR domain-containing adaptor molecule 1 (also known as TRIF), Lipopolysaccharide also activates nuclear factor-κB via myeloid differentiation primary response protein. RNAs activate MDA5 or RIG-I, or double strand DNA activate the DNA sensor cyclic GMP-AMP synthase and then the stimulator of interferon genes protein which all lead to the activation of IRF3. Translocation of IRF3 induces transcription of IFN-β. Translocation of nuclear factor-κB induces transcription of inflammatory cytokines.
As shown in Figure 2, IFN-α and IFN-β exert their immune functions by binding to cell surface receptor IFNAR, causing dimerisation of subunits IFNAR1 and IFNAR2, which activate tyrosine kinase 2 (TYK2) and JAK1 separately. Then, TYK2 and JAK1 phosphorylate STAT1 and STAT2 to form heterodimers, to which IRF9 bind and form the heterotrimeric complex IFN-stimulated gene factor 3. IFN-stimulated gene factor 3 translocates to the nucleus and initiates the transcription of ISGs by combining with IFN-stimulated response elements, producing antiviral and antitumour molecules and transcription factors such as IRFs.
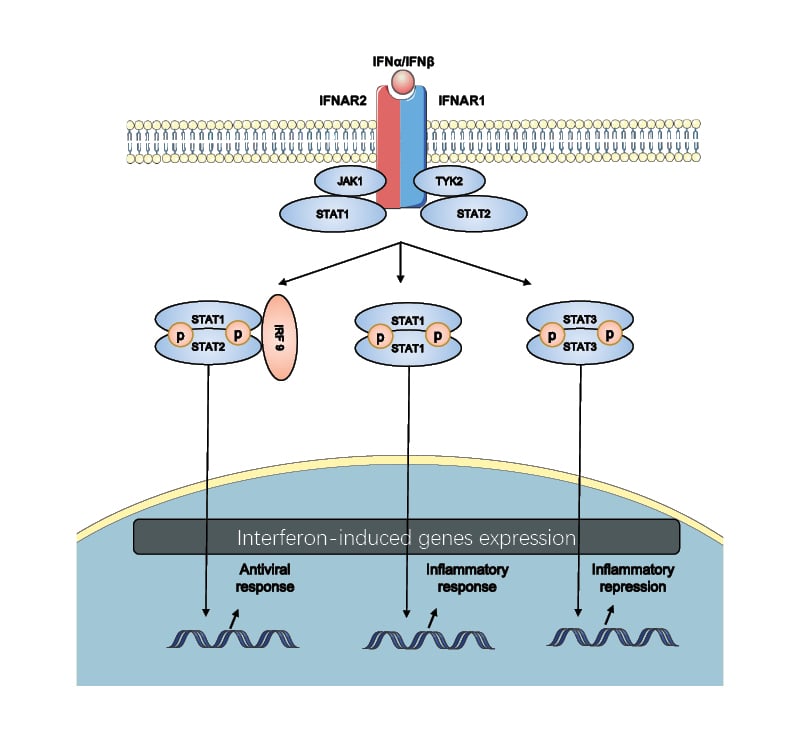
Figure 2: The canonical Type I interferon signalling pathway.
Type I IFNs bind to IFNAR, which is composed of the IFNAR1 and IFNAR2 subunits, initiating a signalling cascade through JAK1 and TYK2. This activates STAT1 and STAT2. STATs get phosphorylated and dimerised and then translocate to the nucleus. Different STATs activate the different sets of genes involved in the biological process, including the antiviral and inflammatory response and inflammatory repression.
IRF9: interferon regulatory factor 9; STAT signal transducer and activator of transcription; TYK2: tyrosine kinase 2.
TYPE I INTERFERON SIGNALLING PATHWAY IN SJÖGREN’S SYNDROME
Studies have shown increased Type I IFNs in SS, correlating with clinical features. Researchers have found that IFN-α was over-expressed in minor salivary glands (MSG), ocular epithelial cells, plasma, and peripheral blood cells in patients with SS.14-17 Nezos et al.18 have found that Type I IFNs and Type II IFNs were overexpressed in peripheral blood and MSG from patients with SS compared with healthy control, while patients with SS and lymphoma had lower IFN-α and higher IFN-γ than patients with SS and without lymphoma. They further showed IFN-α/IFN-γ mRNA ratio in MSG was the best discrimination of lymphoma development in SS.18
Imgenberg-Kreuz et al.19 demonstrated that IFN system activation correlated with adverse prognostic factors including younger age, more positivity of anti-SS-related antigen A (SSA) or SS-related antigen B antibodies, hypergammaglobulinemia, low C4, and lymphoma.19 RNA-containing immune complexes can activate immune cells and produce Type I IFNs. Anti-SSA or SS-related antigen A autoantibodies were specific antibodies for SS that target RNA-binding proteins. They were found to induce Type I IFNs in peripheral blood mononuclear cells in people who are healthy, and correlated with activation of the IFN-I pathway in SS.20
Non-obese diabetic mice spontaneously develop autoimmune inflammation in the lacrimal and salivary glands and are considered a suitable model to study SS.21 Chaly et al.22 have found four upregulated genes related to male-specific lacrimal gland inflammation by gene expression analysis, three of which (CXCL9, CCL19, and EPSTI1) were dependent on Type I IFNs. Furthermore, they found IFNAR1-deficient mice were protected from dacryoadenitis. The results implicated that the Type I IFNs pathway is required for lacrimal gland inflammation in non-obese mice and suggested that the mechanisms of Type I IFNs in human SS.
The source of increased Type I IFNs in circulation and tissues was yet to be known, and many cells were reported to contribute to the process. Maria et al.23 found an increased expression of IFN-I-induced genes in circulating immune cells, which was associated with increased disease activity in SS.23 Considering pDCs are the major source of IFNs-I, Hillen et al.24 performed RNA-sequencing of circulating pDCs from patients with or without SS and showed that SS pDCs produced higher levels of pro-inflammatory cytokines, including Type I IFNs, and Type I IFNs-induced gene signature, which was associated with disease activity. Besides, Lopes et al.25 have performed an RNA-sequencing of monocytes from patients with SS and identified four molecular signatures in monocytes.5 They were related to translation, IFN-signalling, and TLR signalling. Unsupervised hierarchical cluster analysis of the hub-genes identified one cluster characterised by a higher prevalence of anti-SSA antibodies, IFN score, and erythrocyte sedimentation rate, indicating that Type I IFNs altered transcriptional profile of SS-monocytes and participated in the pathogenesis of SS. In another study by Blokland et al.,26 patients with a high Type I IFN signature expressed elevated levels of Fas on innate lymphoid cells (Groups 2 and 3), supporting their role in the pathophysiology of SS.
Intrinsic factors may also contribute to Type I IFNs production. Long interspersed nuclear element 1 is one of the types of endogenous virus-like genomic repeat elements that are silent in normal conditions. It was reported that the expression of long interspersed nuclear element 1 was increased and correlated with the expression of IFN-I in the MSG from patients with SS.27 This may also shed some light on the mechanisms of SS.
To express the activation of IFN-I signalling, the ‘IFN-I signature’ was proposed to describe the increased expression of a variety of IFN-1-regulated genes was observed in peripheral blood cells of patients with systemic lupus erythematosus (SLE).28,29 Later, the IFN signature has also been found in other autoimmune diseases such as SS, myositis, rheumatoid arthritis, and systemic sclerosis.30 Expression of Type I IFNs-induced genes in monocyte from patients with SS was correlated with B cell activating factor and disease activity.31,32 Del Papa et al.33 have found that patients with SS and systemic extra-glandular manifestations have higher Type I-regulated genes versus patients with a disease limited to glandular features. Recently, Cinoku et al.34 have found that SS with lymphoma had higher expression levels of IFNs-I-induced genes, especially ISG15 in both labial MSG and peripheral blood, representing a novel biomarker for lymphoma development in SS. It should be noted that the IFN signature can be induced by either Type I IFNS or Type II IFNs, and it was difficult to differentiate them. Both types of IFNs participate in the pathogenesis and prognosis of SS.18,35 More efforts are needed to develop more specific and sensitive methods to detect both signalling pathways and their distinct roles in SS.
Environmental factors such as viral infections were thought to trigger the development of SS in individuals with a susceptible genetic background.36 Reports of familial aggregation, genome-wide association studies, and candidate gene association studies supported genetic and epigenetic factors of SS.37 The major histocompatibility complex region was the strongest genetic predisposition to SS.38-41 Other than major histocompatibility complex, the two largest genome-wide association studies also identified other polymorphisms that were associated with SS.42,43 The strongest related genes, including IRF5 and STAT4, play a role in the IFN gene signature. STAT4 is a transcription factor in the downstream of Type I IFNs signalling and initiates a transcription of ISGs. IRF5 acts downstream of Type I IFNs and forms a positive feedback loop to induce IFN-α production. However, the exact contribution of these genetic variations to the development of SS is still to be elucidated.
Epigenetic modifications are defined as changes in phenotype without alteration of DNA sequences, consisting of primarily DNA methylation, histone modifications, and non-coding RNAs. Epigenome-wide association studies have found Type I IFN-induced genes in MSG biopsies and, in different cell types, hypomethylation, which correlates with increased mRNA expression in patients with SS.37
MicroRNAs are small single-stranded RNA molecules that can bind to target messenger RNA and interfere with translation. Jang et al.44 evaluated the expression of microRNAs in primary epithelial cells from SS salivary glands and found miR-1248 activated IFN-β through the direct interaction with RIG-I and argonaute 2. Functional studies established two aspects of miR-1248 that affect human salivary glands cells: one served as a ligand to RIG-I and induced IFN production, while the other suppressed the expression of messenger RNAs. Jara et al.45 demonstrated that Type 1 IFNs could decrease the levels of hsa-miR-145-5p in salivary glands of patients with SS, leading to upregulation of Mucin 1 and TLR4, which contribute to salivary gland inflammation and dysfunction in patients with SS. In conclusion, both genetic and epigenetic factors could influence the activation of Type I IFN signalling in SS.
Post-translational modifications also play a role in regulating the Type I IFN signalling pathway.46 Increased histone acetylation of IFN-inducible genes was correlated with their increased expression in autoimmune disease patients.47 More information on the effect of post-translational modifications to Type I IFN pathway need to be explored.
TREATMENT TARGETING THE TYPE I INTERFERON PATHWAY IN SJÖGREN’S SYNDROME
Although many clinical trials targeting the type I IFN pathway have been started, as summarised in Table 1 and Figure 1, few beneficial results have been published. A low dose of orally-administered IFN-α was found to exhibit anti-inflammatory activity through complex immune-mediated effects in patients with SS.58 Successfully treating SS-associated neuropathy with IFN-α was reported in some cases, especially in patients with ganglionopathy.59,60 However, IFN-α administered via the oromucosal route in a combined Phase II clinical trial increased salivary output but failed to meet coprimary endpoints in a combined Phase III study.51,54 As a traditional treatment of SS, hydroxychloroquine is frequently used to treat SS by preventing the activation of TLR7 and TLR9, but failing to improve clinical response.61
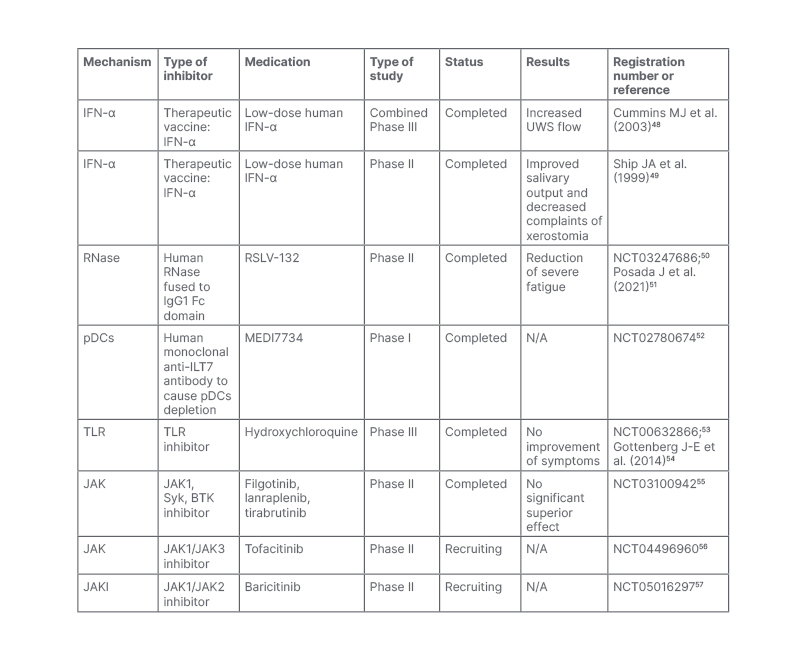
Table 1: Clinical trials of medicine targeting the Type I Interferon pathway in patients with Sjögren’s syndrome.
BTK: Bruton’s tyrosine kinase; IFN: interferon; N/A: not available; pDCs: plasmacytoid dendritic cells; Syk: spleen tyrosine kinase; TLR: toll-like receptor; UWS: unstimulated whole saliva flow.
Blocking Type 1 IFNs is a promising strategy for the treatment of SLE, and, therefore, could also be of interest in SS.62 Anifrolumab, an anti-IFNAR monoclonal antibody, was approved by the U.S. Food and Drug Administration (FDA) on 2nd August 2021 and the European Medicines Agency (EMA) on 21st February 2022 as an add-on therapy for adults with moderate-to-severe SLE.63 It is encouraging news for ongoing clinical trials targeting Type 1 IFNs in treating SLE.
IFN vaccination is successful in preventing SLE progression and improving survival by inducing neutralising anti-IFN-α antibodies in mouse models.64 IFN-Kinoid, an IFN vaccine that can induce a disease-modifying polyclonal anti-IFN-α antibody with high IFN-α neutralisation capacities, was tested in a Phase IIb, randomised placebo-controlled study in adults with SLE with statistically down-regulated IFN signature, though the clinical coprimary endpoint (British Isles Lupus Assessment Group-based Composite Lupus Assessment [BICLA] response with corticosteroids tapering) was not met.65,66 Nonetheless, secondary endpoints including SLE Responder Index (SRL) 4 with corticosteroids tapering and lupus low disease activity state showed superiority in the kinoid group.65 Patients with active virus infection were excluded from the trial regarding an reduction of anti-viral effect of blocking IFN-α. However, no increased risk of viral infections was observed in kinoid group compared with placebo group. One limitation was that kinoid did not block IFN-ω, IFN-β, IFN-γ, or IFN-λ, which may also participate in the development of the disease. IFN kinoid was further used in treating mouse models with SS-like features, resulting in the reduction of Type I IFN signature and disease feature improvement.67 However, there are no clinical trials on anti-IFN-α/IFNAR, and more clinical evidence is needed regarding the safety and efficacy of these drugs in SS.
Circulating RNAs can be sensed by PRR and induce Type I IFN production, RSLV-132, a human RNase fused to human IgG1 Fc domain. They have the ability to digest circulating RNAs and inhibit Type 1 IFN production. It was evaluated in a Phase II randomised double-blind placebo-controlled study with clinically meaningful improvements.51 Larger randomised clinical trials are needed to further confirm its therapeutic effect.
TANK-binding kinase 1 is an important molecule downstream of RIG-I-like receptors, activating IRF3 and IRF7, which results in Type I IFN production. Recently, Bodewes et al.68 demonstrated that TANK-binding kinase inhibition reduced the expression of ISGs in pDCs from patients who tested positive for Type I IFN, representing a potential treatment target in SS.
JAK inhibitors were in the important downstream pathway of Type I IFNs and have exhibited promising results in SLE. Baricitinib, a JAK1/2 inhibitor, has been tested in two double-blind multicentre randomised placebo-controlled Phase III trials for the treatment of SLE. In SLE-BRAVE-I trial (NCT03616912),69 the baricitinib 4 mg oral dose met the primary endpoint for SRI4 response at Week 52 compared with placebo. However, the SLE-BRAVE-II study (NCT03616964),70 which also studied adults with active lupus, neither met the primary endpoint of SRI4 response, nor key secondary endpoints. Considering the similar pathogenesis in SS, studies have been carried out on JAK inhibitors to treat SS. In mouse models, Lee et al.71 demonstrated the filgotinib, a JAK1 selective inhibitor, ameliorated the function of excretion and lymphocytic infiltration of the salivary gland. JAK1/3 inhibitor tofacitinib and JAK1/2 ruxolitinib also exhibited good responses in vitro.72,73 Several interventional clinical trials are on-going and the results are not yet available (Table 1). A randomised Phase II double-blind, placebo-controlled study has assessed the safety and efficacy of JAK1 inhibitor filgotinib, TYK2 kinase inhibitor lanraplenib, and Bruton’s tyrosine kinase inhibitor tirabrutinib in adults with active SS, separately (NCT03100942).55 The primary study results reported on the clinical trials website showed that none of these three drugs were significantly superior to placebo. However, no conclusion can be reached until the full analysis is completed and official papers are published. The other Phase II trials involving tofacitinib and baricitinib were in the recruitment phase (NCT04496960 and NCT05016297).56,57
CONCLUSION
Type I IFN signalling plays a vital role in pathogenesis in SS and provides a promising intervention target for future studies. The sources of Type I IFNs and the induction of IFN signalling in SS still need to be fully elucidated. There are several ongoing clinical trials, and their results are eagerly awaited. Thus, it is of great importance to further explore the role of Type I IFN signalling in SS and other autoimmune diseases.