Meeting Summary
The licensing of biosimilars heralds the start of a new era for physicians treating immune and inflammatory diseases. This symposium provided an update on biosimilar drugs and dealt with questions and concerns around switching from a reference biological drug to its biosimilar.
Prof Isaacs presented the physician’s perspective, describing the regulatory process that is designed to provide reassurance regarding clinical equivalence for biosimilars alongside comparable safety and immunogenicity data. A current consequence of a range of different clinical trial designs is that biosimilars cannot be compared. As more biosimilars enter the market, he made the case for the standardisation of clinical trial designs to simplify comparisons between the different biosimilars. Dr Goll gave an overview of the NOR-SWITCH study. The Norwegian government-funded study showed that switching from reference infliximab (INX) to the biosimilar CT-P13 was not inferior to continued treatment with INX.
Prof Gonçalves shared the pharmacist’s perspective and explained that post-approval pharmacovigilance is crucial for consolidating confidence in biosimilars. He presented studies showing that there was no evidence for biosimilar-related immunogenicity beyond the reference molecule. He concluded that in pharmacovigilance all switching information obtained in registries should be pooled with voluntarily reported and suspected adverse-drug reactions.
Ms Bosworth focussed on the views and needs of patients with regard to key issues associated with switching to biosimilar drugs. She stated honesty and transparency were required when explaining the reasons for switching and that healthcare staff should not hide the fact that saving money is the reason for switching. Financial savings resulting from introducing biosimilars, she stressed, should be shared between commissioners, hospital units, and rheumatology teams. A range of resources on biosimilars for both health professionals and patients are available from the National Rheumatoid Arthritis Society (NRAS).
A New Wave of Biosimilar Drugs
Professor John Isaacs
Welcoming delegates, Prof Isaacs quoted the World Health Organization (WHO)’s definition of biosimilars as a “biotherapeutic product which is similar in terms of quality, safety, and efficacy to an already licensed reference biotherapeutic product”.1 The US Food and Drug Administration (FDA) and European Medicines Agency (EMA), he said, use different definitions for biosimilarity, with the FDA focussing on ‘safety, purity, and potency’ and the EMA ‘quality, safety, and efficacy’. In reality, he said, they are referring to the same concept of using the totality of evidence to confirm bioequivalence of the reference medicine and the biosimilar.
Currently, there is a great deal of activity in the field of biosimilars, with development of biosimilars underway for adalimumab, INX, etanercept, tocilizumab, and rituximab.2 Key issues facing physicians working with this ‘new wave’ of biosimilar drugs, are efficacy and safety (regulation), immunogenicity, pharmacovigilance, extrapolation, working with multiple biosimilars, and switching and substitution.
Biosimilar Studies Show Equivalence
Professor John Isaacs
In comparison with small molecule drugs, biologics represent ‘very sophisticated and complicated molecules’. It is well known that monoclonal antibodies have typical Y-shaped structures, with one end binding antigen and the other having effector functions. What is less well known, said Prof Isaacs, is that many of the antibody functions depend on the fine structure, which can be affected by modifications to the protein sequence. Such modifications (including glycosylation, methylation, deamidation, and oxidation) occur post-translationally, once the protein has been synthesised from RNA.3
As a result, the final monoclonal antibody is not just a product of the DNA/amino acid sequence but ‘critically influenced’ by proprietary factors such as cell lines, the way cells are cultured, and antibodies purified. Just as it may not be possible to make identical bread, wine, and beer (which are also manufactured from living organisms), Prof Isaacs explained, it is not possible to exactly replicate reference drugs.
With biosimilars, Prof Isaacs said, regulators were most interested in whether the drug looks the same as the bio-originator, i.e. whether their structures, including post-translational modifications, are highly similar. They must also behave similarly in in vitro assays, for example, in terms of antigen binding and effector function. Consequently, and in contrast to conventional drugs, regulators require less evidence from clinical studies for biosimilars, and instead place greater emphasis on analytical dossiers. Nonetheless, efficacy and pharmacokinetics (PK) have to be equivalent, and safety and immunogenicity must be shown to be comparable to the reference, with immunogenicity continuing to be assessed post-marketing. Provided the above criteria are met, there is no requirement for formal efficacy trials of biosimilars; if similarity is established with the bio-originator, it is assumed that the biosimilar will be effective in the same range of indications.4
Prof Issacs provided the example of CT-P13 development, an INX biosimilar that was the first biosimilar to be licensed by the EMA. The Phase I PLANETAS study5 showed that, at the fifth dose, the geometric mean area under the concentration-time curve was 32765.8 µgh/mL for CT-P13 versus 31359.3 µgh/mL for INX with a ratio of geometric means of 104.5%. Furthermore, the serum Cmax geometric mean was 147.0 for CT-P13 versus 144.8 for INX (ratio of geometric means of 101.5%). Both of these measures fell within the pre-defined equivalence margins.
In the Phase III efficacy study, at Week 30, the American College of Rheumatology (ACR) Criteria ACR20 responses were 60.9% for CT-P13 versus 58.6% for INX in the intention-to-treat population response rate, giving a treatment difference of 2% (95% confidence interval [CI]: -6%, 10%), again well within the equivalence margin for the trial.6
Extrapolation describes the situation where a biosimilar which was considered equivalent in one indication is also considered to work in a second indication (without demonstrating this in an additional clinical trial).7 To illustrate the concept, Prof Isaacs gave an example. If a biosimilar is equivalent to the reference medicine in disease X, and if the reference is effective in diseases X, Y, and Z, then the biosimilar can be considered effective in all three conditions. In fact, the reference clinical trial data becomes incorporated into the biosimilar summary of product characteristics.
It should be noted that, for extrapolation to be accepted, therapeutic efficacy must rely on a similar mechanism of action for both the original indication where the confirmatory study was conducted in and the extrapolated indication, e.g. INX in rheumatoid arthritis and psoriatic arthritis.
Switching and substitution, said Prof Isaacs, represented one of the most controversial topics. Switching, which refers to a clinician deciding to change from a reference to a biosimilar, represented a very different situation from substitution, which involves a third party (such as a pharmacist) making that decision. This is not currently allowed anywhere in the world, although the FDA are considering a category of ‘interchangeability’ that would allow substitution, subject to rigorous clinical trial data involving multiple switches between reference and biosimilar.
For pharmacovigilance, product identification is of paramount importance. In Europe, biosimilars use the same international non-proprietary name e.g. INX, while in the USA they are given distinct names with the international non-proprietary name modified by a suffix (e.g. etanerceptszzs, INX-dyab). Prof Isaacs noted the danger that the European system could inappropriately suggest interchangeability, underlining the need to use brand names within Europe. He provided an overview of the different trial designs used in biosimilar switching studies to confirm clinical bioequivalence for biosimilar approval (Figure 1).2 The difference in design does not allow for direct comparison of efficacy and safety data of different biosimilars.
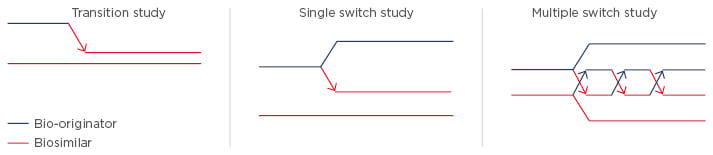
Figure 1: Examples of biosimilar switching studies.2
Transition Studies
In a transition study, half the subjects receive reference drug and half the subjects receive biosimilar, and the reference group is subsequently switched to the biosimilar. The idea is to address whether the transition has, for example, precipitated immunogenicity. Examples of transition studies are PLANETRA and PLANETAS, which compare CT-P13 and INX.5,6,8,9
Single Switch Studies
In a single switch study, half the patients receive the reference and half receive the biosimilar, and then half the reference group is switched to the biosimilar and half remains on the same treatment. This approach allows comparison of the two arms to see if there are any differences in outcome. An example is the ABP 501 study exploring an adalimumab biosimilar in psoriasis.10
Multiple Switch Studies
Multiple switch studies involve switching therapies multiple times (alternating) between the reference drug and a biosimilar. The objective is to look at the PK after each switch, as well as efficacy and immunogenicity. An example of a multiple switch study is EGALITY comparing the etanercept biosimilar GP2015 with its reference in psoriasis.11
With a future prospect of multiple biosimilars, Prof Isaacs questioned what would happen if formularies were to subsequently adopt multiple biosimilars (A, B, C, D, or E) and whether physicians could feel confident switching between these biosimilars and back to reference. At present, a difficulty is that biosimilar trial designs are heterogeneous, he said, making indirect comparisons problematic.
In a recent editorial that he wrote with Jonathan Kay,4 Prof Isaacs argued that as more biosimilars enter the market, standardisation of clinical trial design would allow for indirect comparisons between them, adding confidence to decision-making around switching. Standardisation, they suggested, might include studying healthy subjects versus patients (in Phase I), specific inclusion and exclusion criteria, equivalence margins, primary endpoints (including timing of assessment), secondary endpoints (including timing of assessment), PK assays (endpoints compared and timing of assessment), immunogenicity (assays used and timing of testing), analysis of effects of immunogenicity on PK, efficacy and safety, definition of adverse events, statistical analyses, and crossover designs beyond the primary endpoint (in Phase III). If adopted, standardisation would simplify indirect comparison between biosimilars and help clinicians to feel more comfortable about switching.
Clinical Evidence for Biosimilars and Switching: The Nor-Switch Study
Doctor Guro Løvik Goll
Dr Goll gave a lucid introduction into the Norwegian healthcare system, where 100% of healthcare costs are covered by the government. With increasing use of biologics over the past 7 or 8 years, pharma companies in Norway have been invited to tender for providing treatments, with cost calculations proving a key consideration for choosing the winning options. It was important to stress that the tendering process only applies to patients starting their first biologic, and furthermore, if physicians feel there are ‘good reasons’ to choose different drugs they can make a special case for individual patients.
A substantial discount in the price of biosimilar INX in early 2015 led to a dramatic increase in its use in Norway and highlighted the key clinical question of what to do with patients who were already stable on the reference biologic Remicade® (INX). Questions included whether it was safe or even ethical to switch them to the biosimilar? The NOR-SWITCH study,12 funded by the Norwegian government, set out to assess if CT-P13 was non-inferior to innovator INX with regard to disease worsening in patients who had been on stable INX. The study took place across 40 centres in Norway, involving 16 rheumatology departments, 19 gastroenterology departments, and 5 dermatology departments.
Inclusion criteria for NOR-SWITCH were clinical diagnoses of either rheumatoid arthritis (n=78), spondyloarthritis (n=91), psoriatic arthritis (n=30), ulcerative colitis (n=93), Crohn’s disease (n=155), or chronic plaque psoriasis (n=35). To be eligible, patients needed to have undergone stable treatment with INX for the last 6 months.
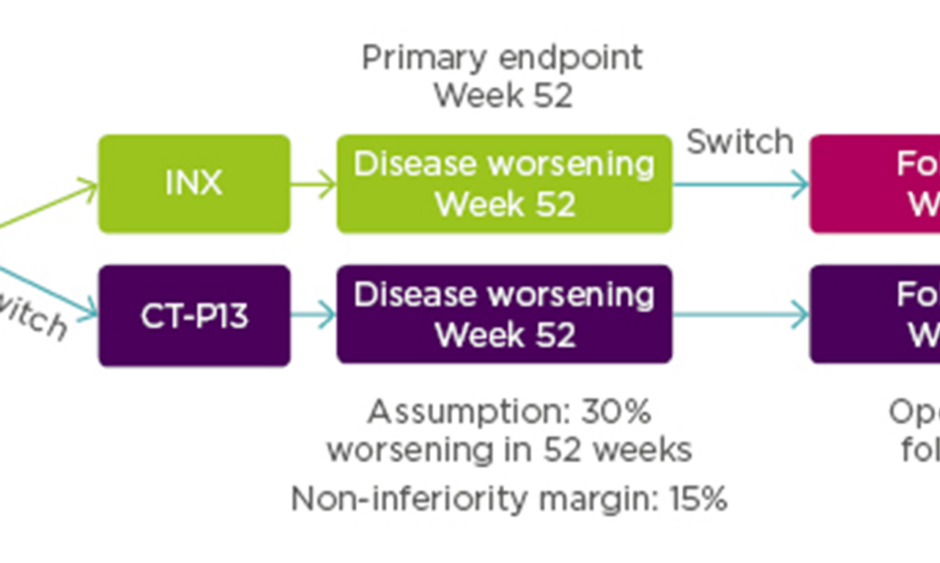
Enrolled patients (N=482) were randomised 1:1 to continue treatment with INX (n=241) or to be switched from INX to CT-P13 (n=241). Additionally, 380 patients from both arms were entered into an open label extension study with all patients treated with CT-P13. For this group additional assessment was scheduled for Week 78. See Figure 2 for the NOR-SWITCH study design.
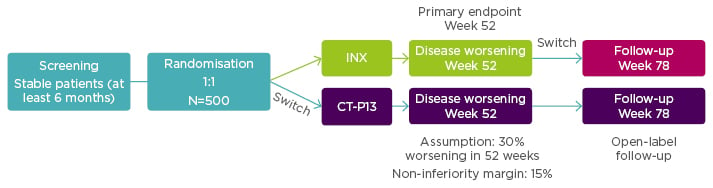
Figure 2: NOR-SWITCH study design.12
A randomised, double-blind, parallel-group study to evaluate the safety and efficacy of switching from innovator infliximab to biosimilar infliximab compared with continued treatment with innovator infliximab in patients with rheumatoid arthritis, spondyloarthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and chronic plaque psoriasis.
INX: infliximab.
From power calculations, using a 15% non-inferiority margin with 30% disease worsening at Week 48 (based on a power of 90% and alpha 2.5%) it had been estimated that the NOR-SWITCH study would need to treat 394 patients.
Results showed the primary endpoint (disease worsening assessed at Week 52 using different definitions for each of the six diseases) occurred in 26.2% of patients continued on INX compared to 29.6% switched to CT-P13 (rate difference: -4.4%; 95% CI: -12.7 to -3.9). Although results fell within the 15% prespecified non-inferiority margin for CT-P13 to INX, data for Crohn’s disease fell close to the non-inferiority margin. Dr Goll said that, investigators have been cautious about drawing conclusions based on the individual diagnoses, and it was thought likely that Crohn’s disease was a ‘spurious’ finding since they did not see any signals for C-reactive protein or disease remission. Serum drug trough levels for INX and CT-P13 were similar from baseline to Week 52. Additionally, incidence of anti-drug antibodies occurred in 7.1% of patients taking INX versus 7.9% taking CT-P13.
The strengths of NOR-SWITCH, said Dr Goll, included the randomised controlled trial design, comprehensive data collection, inclusion of sufficient numbers of patients according to power calculations, having patient representatives in the project group, government finance (no industry involvement), and that drugs were provided through the regular payment schedule. Limitations included the finding that it was not powered for non-inferiority within each diagnostic group and the absence of data on patients who declined participation.
The NOR-SWITCH trial, said Dr Goll, showed that switching from INX to CT-P13 was not inferior to continued INX treatment and supported switching from INX to CT-P13 for non-medical reasons.
Safety and Immunogenicity in Switching
Professor João Gonçalves
Currently, said Prof Gonçalves, there are around 50 biosimilars in clinical development. Biosimilars, he explained, have stringent assessment criteria that include clinical trials and functional and analytical testing.
The goal for companies is to show consistent quality, which in turn delivers consistent safety and efficacy. It was important to pay close attention to critical quality and safety variables including:1
- Immunogenicity (aggregates, impurities)
- Safety/toxicity (antibody purity, antibody glycosylation, antibody modifications)
- PK (antibody structure, antibody glycosylation)
- Efficacy (antibody glycosylation mechanism of action in all indications)
For biosimilars, extrapolation to other unstudied indications is possible, based on all data generated with the biosimilar (totality of evidence concept). For example, biosimilar INX CT-P13 was first studied in rheumatoid arthritis and ankylosing spondylitis, but data have been extrapolated to allow use in psoriatic arthritis, rheumatoid arthritis, ulcerative colitis, Crohn’s disease, psoriasis, and ankylosing spondylitis. Prof Gonçalves gave the example of seven approved biosimilars studied in seven indications and then used in 22 different indications.14
Data from Prof Gonçalves’ laboratory (unpublished) found fewer adverse events for CT-P13 when the number of adverse events occurring in the first 3 years after the launch of INX were compared to the number of adverse events occurring in the first 3 years after launch of CT-P13.15 What is crucial for consolidating confidence in biosimilars, said Prof Gonçalves, is continuation of active pharmacovigilance after biosimilars come to market.
The uncomfortable reality of all biological treatments is that manufacturing changes happen frequently, which can result in structure and function differences. It is important that regulators and companies know how to manage such variability, said Prof Gonçalves. ‘Critical quality attribute’ testing can be used to ensure biological drugs act in similar ways to previous batches. He explained that safety issues more often arise with the reference medicine than in the biosimilar.
A further concern is the immunogenic sensitivity of inflammatory diseases, which are sensitive to aggregated proteins. Therapeutic proteins have a propensity for aggregation (during manufacture, shipping, and storage) and the presence of aggregates may induce adverse immune responses in patients that may affect safety and efficacy.16 Aggregates are believed to result in amplification of anti-drug immune responses including enhancing T cell responses and activation of dendritic cells.
The risk of anti-drug immune responses is known, said Prof Gonçalves, and its main consequence is that patients who are anti-drug antibody positive might achieve lower drug response rates. One meta-analysis assessing the effects of anti-drug antibodies on response to INX, adalimumab, and etanercept showed that patients with rheumatoid arthritis, spondyloarthritis, psoriasis, and inflammatory bowel disease who were antidrug antibody positive achieved lower response rates than patients who were anti-drug antibody negative (risk ratio: 0.32; 95% CI: 0.22 to -0.48).17
The probability of developing immunogenicity does not depend on the drug alone. A number of factors influence the probability of immunogenicity.18,19 Those associated with the product include whether self or non-self, the presence of T cell epitopes, formulation (including impurities and aggregates), and post-translational modifications. Those not associated with the product include routes of administration (intravenous versus subcutaneous), whether used for an acute or chronic disease, PK, and whether the target was cellular or soluble. Finally, factors associated with patients include haplotype, tolerance to protein, immunosuppression, and pathology.
An interesting question, said Prof Gonçalves, was whether the immune system identifies the biosimilar as different from the reference medicine. The factor that is often forgotten, he said, was that biosimilars and reference biologic have exactly the same amino acid sequences leading to low risks of B cell activation. It would only be triggered by protein aggregation. Recently, when Prof Gonçalves and colleagues undertook a cross reactivity assessment between CT-P13 and IFX using anti-CT-P13 patient sera, they showed strong correlations suggesting no evidence of new epitopes in CT-P13.20 (Figure 3)
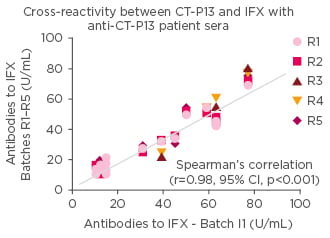
Figure 3: Studies showing that the immunogenic epitopes show no difference between CT-P13 and infliximab.
No evidence of new epitopes in biosimilar IFX. IFX: infliximab; CI: confidence interval.
When investigators went on to identify every monoclonal antibody produced by patients for both the original drug and biosimilar, they found no differences between the Fc and Fab regions. Such findings, said Prof Gonçalves, suggest the immune system recognises the two drugs in the same way. A range of studies exploring immunogenicity in INX, adalimumab, and etanercept biosimilars showed that the risk of switching was minimal for both switching to biosimilars and also using different biosimilar batches.8,21-23
Emphasising the importance of pharmacovigilance around immunogenicity for both original biologics and biosimilars, Prof Gonçalves said measures should be introduced to ensure traceability of batches and products. He suggested there should also be possibilities to define and monitor clinical endpoints relevant to potential risk of immunogenicity.24
It was important, he added, to establish immunogenicity endpoints that would be measured during a managed biosimilar switch. They could include drug trough levels and immunogenicity testing, adverse events (which could be included in a biologic registry), patient-reported side effects, patient-reported outcome measures, disease activity assessments, laboratory tests of inflammation (C-reactive protein and erythrocyte sedimentation rates), other blood tests, and economic endpoints. The frequency of follow-up and the member of the healthcare team administering the test would also need to be decided.
An integrated approach is needed. Prof Gonçalves added that all switching information obtained from registries should be pooled with voluntarily reported and suspected adverse drug reactions in pharmacovigilance. Furthermore, to eliminate drug-related causes of concerns the pharmacy should undertake a risk assessment model to consider unsafe handling of biological agents that could lead to altered immunogenicity.
The Patient’s Voice in Biosimilar Use and Switching Decisions
Ms Ailsa Bosworth
Patients, said Ms Bosworth, require reassurance and explanations about the differences between biologics and biosimilars in language and terminology that makes sense to them. The experience of the NRAS has been that patients accept switching when reasons are explained to them in ‘accessible’ ways that they can relate to.
Honesty, said Ms Bosworth, is required when explaining reasons for switching. Health staff should not hide the fact that saving money is the reason for switching. In economies where health systems are under strain from ageing populations it is right to be careful about use of resources. However, Ms Bosworth said, she had come across examples of units saying that switching would mean that more patients would benefit from new treatments at earlier stages in their disease pathway. In the UK, she said, this was not the case since the National Institute for Health and Care Excellence (NICE) had strict eligibility criteria. NRAS, she said, has just launched a new booklet, ‘Medicines in Rheumatoid Arthritis’, covering such issues.
When considering switching, not all patients are suitable candidates. Ms Bosworth said that patients who do well on biologics will most probably do well with biosimilars, but patients who have reacted badly to biologics or had difficulty becoming stable may not be suitable to switch. Healthcare teams must review their patients carefully before making decisions to ‘switch all’.
NRAS believes when decisions are taken to switch patients to biosimilars, that stakeholder groups should be established for discussions around the switch programmes with representatives from all parties involved, including at least two patients. The rationale for including two patients, she explained, was to prevent individuals from feeling isolated. Sadly, there are examples of switching programmes that have been implemented without consulting patients. In the UK, patients are most often advised by letter that they are going to be switched. This, said Ms Bosworth, can work if such letters are accurate, appropriately worded, and give patients the opportunity to contact members of their healthcare teams to discuss any concerns.
NRAS resources on biosimilars include its position paper, a video interview with the NRAS Chief Medical Advisor Prof Peter Taylor, an NHS England publication “What is a Biosimilar”, a stakeholder review, and a report of the NHS England Biosimilar Medicines Workshop. NRAS, said Ms Bosworth, is happy to advise units on the wording for patients’ letters about switching.
Ms Bosworth explained that data collection should be considered important by everyone, with manufacturers investing in registries and patients providing data for them. Registry data on biologics collected in registries across Europe has provided enormous reassurance around safety. Now, she said, it is vital to collect similar data on biosimilars. While currently the numbers of patients on biosimilars are most likely insufficient to identify rare side effects, the increasing number of patients who will in future be prescribed biosimilars should enable this.
When it comes to sharing the savings resulting from switching patients, NRAS believes they should be split in an equitable way between the commissioners, the hospital units, and the rheumatology teams. When rheumatology teams (who implement the work of switching) receive a share they can invest in patient services. In the UK, there have been examples where rheumatology teams have invested their share of savings into appointing additional nurse specialists. However, Ms Bosworth cautioned that in the UK there have been delays lasting >12 months while stakeholders argue about who receives the savings. It was important, she stressed, to settle such discussions quickly since as prices shift downwards the current levels of gain will not last.
To illustrate comprehensible educational materials in patient-appropriate language as provided by NRAS, Ms Bosworth closed her presentation with a clip from an educational interview on the issues of biosimilars she had undertaken with NRAS’ Chief Medical Advisor Prof Peter Taylor.25 Patient organisations are partners of choice for patients as well as healthcare professionals to optimise the shared decision-making process in biosimilar use and switching.