Abstract
Oedema is a hallmark feature of nephrotic syndrome (NS) and can cause significant patient morbidity. The pathogenesis of oedema formation is complex and results from abnormalities in sodium retention, inter-play of neurohormonal factors, and changes in capillary filtration barrier. Salt retention is often primary (‘overfill’ theory) because of increased sodium-potassium adenosine triphosphatase activity in the collecting duct cells, increased direct epithelial sodium channel activation (ENaC) by urinary proteases (independent of aldosterone), and an overall increased effective arterial blood volume. However, a subset of patients with NS, especially children, demonstrate decreased effective arterial blood volume (‘underfill’ theory) and secondary sodium retention as the primary mechanism of oedema formation. Increased capillary permeability and vascular inflammation contributes as well. Loop diuretics with or without salt-poor albumin are the mainstay of therapy in adults, although no large clinical trials exist to guide diuretic choice or dosage. Combination diuretic therapy is recommended to achieve multi-site nephron blockade and overcome diuretic resistance, which is a frequent challenge. Use of direct ENaC inhibitors (amiloride) in combination with loop diuretics may be especially beneficial given the primary role of ENaC in sodium retention. Aquaretics such as vasopressin receptor antagonists may have a role in treatment as well. Well-designed clinical trials are essential to guide therapy of refractory oedema in NS. In this review, the authors discuss the pathogenesis of oedema formation in patients with NS and propose a treatment algorithm for management of resistant oedema based on the limited available evidence.
INTRODUCTION
Nephrotic syndrome (NS) is characterised by hypoalbuminaemia, heavy proteinuria (>3.5 g/day in adults and urinary protein to creatinine ratio ≥2 mg/mg in children), hyperlipidaemia, and oedema and indicates glomerular pathology. The cause of NS can be idiopathic or secondary to other disorders, and specific treatment is determined by histopathology and underlying aetiology. Interstitial oedema (and in its severest form, anasarca) is a hallmark feature of NS and results in significant patient morbidity, restricted mobility, discomfort, and increased risk of skin and soft tissue infections. Some patients may accumulate up to 30% of extra body weight.1 Oedema results from abnormal sodium (Na+) retention, inter-play of neurohormonal factors, and changes in capillary filtration barrier. Understanding of the pathophysiology of oedema formation in NS has evolved over the years, from the initial ‘underfill’ and subsequent ‘overfill’ theory to recent insights into the role of vascular hyperpermeability and direct epithelial Na channel (ENaC) activation. Treatment of oedema can be challenging because of multiple contributing factors, variability in volume status, and issues with diuretic resistance. Diuretics are frequently necessary for treatment of severe oedema; however, data guiding oedema treatment remains limited and unsupported by large clinical trials. In this review, the authors aim to highlight important theories pertaining to the pathophysiology of oedema formation in patients with NS and treatment strategies for associated refractory oedema.
THE UNDERFILL THEORY OF OEDEMA FORMATION: SECONDARY SALT RETENTION
The ‘underfill’ theory (Figure 1) provides one of the earliest explanations of oedema formation in patients with heavy albuminuria and proposes that, despite the presence of oedema, patients with NS are in fact volume depleted.2,3 It suggests that decreased plasma oncotic pressure due to hypoalbuminaemia alters capillary Starling forces, resulting in extravasation of intravascular fluid into the interstitial space and a decrease in effective arterial blood volume (EABV).1 In an attempt to restore EABV, there is compensatory activation of the renin–angiotensin–aldosterone system (RAAS) and other neurohormonal responses (e.g., anti-diuretic hormone [ADH], angiotensin II [AgII]) leading to secondary Na+ and water retention and oedema formation.
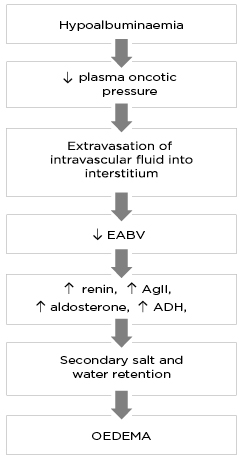
Figure 1: The pathophysiology of oedema formation according to the ‘underfill’ theory.
ADH: anti-diuretic hormone; AgII: angiotensin II; EABV: effective arterial blood volume.”
There are several studies that support the underfill theory. Adult patients with NS with severe hypoalbuminaemia (<1.7 g/dL) have been shown to have evidence of low EABV (low atrial natriuretic peptide [ANP], elevated AgII, and plasma renin activity [PRA], with increased proximal tubular Na+ reabsorption) compared to those with serum albumin >1.7 g/dL. This suggests that severity of hypoalbuminaemia correlates with hypovolaemia and upregulation of compensatory Na+ retaining mechanisms, which is reversed by volume expansion.4 Elevated serum ADH, high PRA, impaired free water excretion, and hyponatraemia are also noted in patients with NS.5 Several other studies6,7 provide evidence that hypovolaemia triggers neurohormonal changes (increased RAAS, ADH, and AgII), resulting in secondary Na+ and water retention in NS. The specific role of aldosterone in facilitating Na+ retention in NS was highlighted in a study of 5 patients with NS and 6 controls given a high-salt diet for 8 days. After 4 days of high salt intake, the control group was able to maintain salt balance while the nephrotic group demonstrated positive Na+ balance. Spironolactone was administered on Day 5. After 72 and 96 hours of spironolactone initiation, patients with NS exhibited a significant increase in natriuresis (205±20 versus 312±13 mEq/day; p<0.005) compared to control subjects, suggesting that aldosterone contributes to salt retention in NS.8
CONTROVERSIES AROUND THE UNDERFILL THEORY
Although the underfill theory provides a plausible conceptual framework for oedema formation in NS and is backed by observational data demonstrating hypovolaemia as an initial precipitant, several other studies have found contrary evidence of expanded EABV and suppressed RAAS in patients with NS, raising concerns about the validity of the underfill theory.9 The role of hypoalbuminaemia as a trigger has also been questioned.10
A key study in paediatric patients described the heterogenous clinical presentation in NS with regards to volume and neurohormonal status.11 The authors evaluated paediatric patients with a history of steroid-responsive NS, who showed signs of relapse detected by home urine dipstick screen, and divided them into three groups based on clinical presentation: proteinuric patients without hypoalbuminaemia; patients with severe hypoalbuminaemia, oedema, and hypovolaemia (high plasma renin, aldosterone, and serum norepinephrine; low ANP and estimated glomerular filtration rate); and patients with severe hypoalbuminaemia and oedema similar to the previous group but without biochemical or clinical evidence of hypovolaemia. Interestingly, the first group demonstrated oedema and slightly increased aldosterone levels despite normal serum protein levels and lack of hypovolaemia. This study not only highlights the variability in clinical features, EABV, and neurohormonal responses in oedematous patients with NS, but was also one of the first studies to suggest that salt retention may precede overt hypoalbuminaemia, questioning the underfill theory. Several studies confirm that a significant number of patients with NS are in fact volume expanded.12,13 Geers et al.12 used 131I-albumin to determine plasma volume and calculated blood volume in 88 adult patients with NS and found that both were either normal or slightly elevated. Another study using 131I-albumin in adult oedematous patients with NS showed that at least two-thirds had either normal or elevated blood volume compared to healthy controls.13 Furthermore, studies employing blockade of the RAAS axis using angiotensin-converting enzyme inhibition were unable to demonstrate increased natriuresis in adults with NS, questioning the role of RAAS in oedema formation.14
The central role of hypoalbuminaemia in oedema formation has also been questioned.10 Patients with congenital defects of albumin synthesis do not develop overt oedema despite analbuminaemia.15 Nagase analbuminaemic rats are able to maintain transcapillary oncotic pressure without signs of abnormal volume regulation.16 A study looking at 62 patients with NS found no correlation between serum albumin levels and blood volume or PRA.17 There is evidence that patients with NS are in fact able to regulate their plasma volume fairly well despite hypoproteinaemia. Koomans et al.18 showed that patients with NS with normal renal function were able to maintain normal intravascular blood volume, even at significantly expanded extracellular fluid volumes. The lack of oedema despite modest hypoalbuminaemia is explained by the parallel drop in interstitial albumin concentration and thus interstitial oncotic pressure, with slight increase in interstitial hydrostatic pressure, counteracting oedema formation. This occurs due to an increase in lymphatic drainage.19,20 Furthermore, correction of hypoalbuminaemia via an albumin infusion is insufficient to stimulate natriuresis and correct oedema. Studies in paediatric patients demonstrate diuresis and improvement in oedema with initiation of steroid therapy, even before the resolution of hypoalbuminaemia, suggesting that factors beyond serum albumin levels determine oedema formation in NS.21
THEORY OF PRIMARY SALT RETENTION: THE OVERFILLING THEORY
The ‘overfill’ theory (Figure 2) describes Na+ retention and expanded EABV as the primary process in oedema formation (and not secondary to hypovolaemia), with onset well before the development of severe proteinuria and hypoalbuminaemia. This model is supported by evidence that a significant subset of patients with NS are volume expanded with suppressed RAAS, as discussed above.
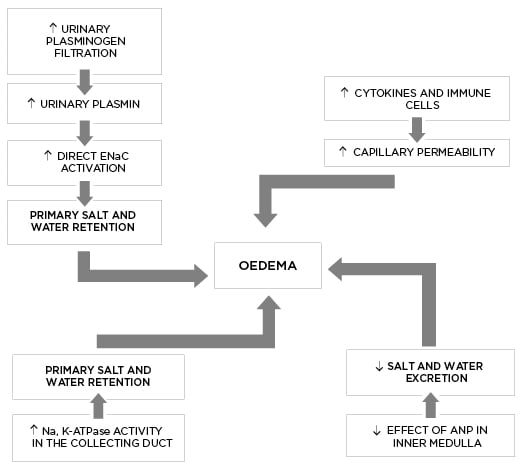
Figure 2: The pathophysiology of oedema formation according to the ‘overfill’ theory.
ANP: atrial natriuretic peptide; ENaC: epithelial sodium channel; Na,K-ATPase: sodium-potassium adenosine triphosphatase.
Current understanding of the site and mechanism of Na+ retention in NS is derived from experimental animal models of disease. The puromycin amino-nucleoside (PAN) rat model of minimal change disease demonstrated that, after a single injection of PAN, these animals developed Na+ retention and massive proteinuria 2–3 days and 4–5 days later, respectively.22 Using micro-puncture techniques in a rat model of unilateral PAN NS, the aldosterone-responsive connecting tubule and the cortical collecting duct (CCD) were isolated as the primary sites of Na+ retention.23,24 However, the role of aldosterone itself in mediating salt retention in PAN nephrotic animals and NS is questionable. In the unilateral PAN nephrotic model, Na+ retention was abolished by amiloride but not by aldosterone receptor blockade, indicating that transepithelial Na+ absorption was independent of aldosterone.24 In another study, bilateral adrenalectomised PAN nephrotic rats received supplemental infusion of physiologic aldosterone and glucocorticoid.25 Despite the inability to increase aldosterone production, these animals demonstrated Na+ retention and volume expansion similar to those with intact adrenals. The primary mechanism was described to be an increase in activity of the basolateral sodium-potassium adenosine triphosphatase (Na+,K+ ATPase) channels in the principal cells of the CCD, due to increased transcriptional induction of the α and β subunits and insertion of new Na+,K+-ATPase pumps. Additionally, increased ENaC activity was seen, which was attributed to aldosterone-induced mobilisation of pre-existing pool of sequestered intracellular channels to the apical membrane of the principal cells rather than an increase in transcription. The study concluded that the induction of Na+,K+-ATPase channels in the CCD is independent of aldosterone and is a robust process that drives ENaC-mediated Na+ retention in PAN nephrotic rats.25
Recent data suggest that proteinuria itself may drive primary salt retention via direct ENaC activation, independent of systemic volume or hormonal triggers. Proteolysis of the ENaC α and γ subunits via proteases (such as plasmin, trypsin, kallikrein, channel-activating proteases etc.) is an important physiologic mechanism of ENaC activation. Normal urine contains small amounts of proteases; however, there is a substantial increase in their filtered load in nephrotic urine.25-27 Inappropriate ENaC activation by proteases can result in pathologic salt retention and oedema in NS.26 There is reasonable evidence that filtered inactive plasminogen in nephrotic urine is converted to plasmin by tubular urokinase-type plasminogen activator.26 Activated plasmin further mediates proteolysis of the γ-subunit of ENaC, resulting in its activation. Several studies confirm the role of proteolytic ENaC activation in pathogenesis of salt retention. Urine from nephrotic humans and animals has been found to activate ENaC currents in vitro.27 Pharmacologic inhibition of urinary protease using aprotinin in nephrotic animals prevented volume retention by inhibiting proteolytic ENaC activation.28 A recent study in 203 adult patients with NS identified urinary plasminogen-plasmin to creatinine ratio as an independent risk factor for oedema formation, and a post-treatment decrease in the ratio as an independent predictor of oedema remission, supporting the role of plasmin-dependent ENaC activation as an important mechanism of salt retention.29 Amiloride, an ENaC blocker, has been shown to ameliorate salt retention and improve natriuresis in various animal and human studies of NS.28,30,31 Interestingly, its effectiveness extends beyond just ENaC blockade. In a podocin gene knockout mouse model of NS, intraperitoneal administration of amiloride was associated with a reversible and significant decline in conversion of urinary plasminogen to active plasmin.30 Amiloride treatment is associated with decreased urine urokinase-type plasminogen activator activity.30,31 Thus, amiloride may attenuate Na+ retention in NS by several mechanisms.
Several endocrine and paracrine regulators of Na+ excretion have been evaluated in NS. ADH,32 insulin-like growth factor-1,33,34 TNF-α,35 thiazolidinedione agonists of peroxisome proliferator-activated receptor γ,36 AgII,37 and nitric oxide38 have all been shown to enhance Na+ retention independent of aldosterone. However, in the PAN nephrotic model, blocking the activity of each of these factors, except AgII, did not show any improvement in natriuresis. Interestingly, increased natriuresis was observed in patients with NS treated with irbesartan for angiotensin-1 receptor blockade but Na+ excretion was modest and short-lasting.1 Perhaps the aldosterone-independent effect of AgII is responsible for the ENaC-mediated Na+ retention only to a limited extent. In addition, patients with NS may have increased sympathetic activity and elevated ADH and ANP levels but decreased responsiveness to their diuretic and natriuretic effects, respectively.39,40 In a rat model, renal sympathetic denervation improved excretion of infused normal saline as well as response to ANP infusion.40 Additionally, Rodríguez-Iturbe B et al. postulated that nephrotic conditions with increased inflammatory infiltrate were more likely to develop Na+ retention. Conversely, children with minimal change disease and no interstitial infiltrate are less likely to have Na+ retention and more likely to belong to the cohort of patients with hypovolaemia, suggesting the role of paracrine inflammatory mediators in Na+ retention.41
INCREASED VASCULAR PERMEABILITY
In addition to primary Na+ retention, there is reasonable evidence that patients with primary nephrotic glomerulopathies have capillary hyperpermeability, which contributes to the development of interstitial oedema. Rostoker et al.42 found significantly high capillary permeability (measured using technetium-labelled albumin) in patients with NS and those with idiopathic cyclic oedema (a condition of abnormal vascular permeability) compared to healthy controls. Interestingly, vascular hyperpermeability improved after treatment with steroids in those with minimal change disease. The proposed mechanism of vascular hyperpermeability was the likely presence of a vascular permeability factor released by local immune cells, although this has not been definitively identified yet.
TREATMENT OF NEPHROTIC OEDEMA
Oedema treatment in NS should be tailored to the individual patient depending on the more dominant underlying pathophysiology (overfill versus underfill). In the overfilled state, diuretics are the mainstay of therapy.44 Despite the presence of interstitial oedema, patients with underfill pathophysiology and low EABV may require initial volume expansion with salt-poor albumin prior to initiating diuretics to minimise diuretic-induced pre-renal acute kidney injury. Therefore, an important initial step in the management of oedema in NS is a determination of EABV using supportive clinical parameters such as orthostatic hypotension, pulse rate, haematocrit, PRA, serum aldosterone, capillary refill time, urine output, blood urea nitrogen–creatinine ratio, and urinary electrolytes.45 In paediatric patients with NS, a fractional excretion (Na+)<0.5 and ratio of Urine (K+) to the sum of Urine (Na+) and Urine (K+) >0.6 may suggest hypovolaemia.46,47 In addition, cardiothoracic index in children (determined by a chest X-ray) or echocardiographic parameters (inferior vena cava index, inferior vena cava collapsibility index, and left atrium diameter) can be helpful.48 These physical and biochemical parameters are not specific to NS but may provide supportive clues to the underlying volume status.
GENERAL MEASURES
Dietary salt restriction (<35mg Na+/kg/day), use of compression stockings, adequate nutrition, limb elevation, and avoidance of non-steroidal anti-inflammatory drugs are important general measures to treat oedema in NS.
Non-steroidal anti-inflammatory drug-induced intrarenal prostaglandin inhibition leads to Na+ and water retention and a decrease in afferent arteriolar dilation, leading to worsening hypervolaemia and reduction in glomerular filtration rate, respectively. Patients with NS are particularly sensitive to these deleterious effects.49 In children with mild, asymptomatic oedema, conservative management with fluid restriction to two-thirds of maintenance and salt restriction is often enough, especially when dealing with steroid-sensitive NS which responds within 4–8 days after initiation of steroids.50
DIURETIC THERAPY
Diuretics are often necessary for treatment of oedema in adults with NS, either alone or with salt-poor albumin. In the paediatric population, diuretic use is reserved for patients with moderate and severe oedema (defined by weight gain of 7–10% or >10%, respectively), those with life-threatening complications such as massive pleural effusions, pulmonary oedema, or cases of steroid-resistant NS with close monitoring for diuretic-induced hypovolaemia.51 Judicious use of diuretics is recommended after confirmation of volume status as the use of diuretics in the underfilled cohort raises risk of hypovolaemic shock and thrombotic events.
Unfortunately, there are no established guidelines to help determine optimal diuretic class or dosage in patients with NS and there is significant heterogeneity in management. Based on the limited data available, the authors propose a treatment algorithm (Figure 3).
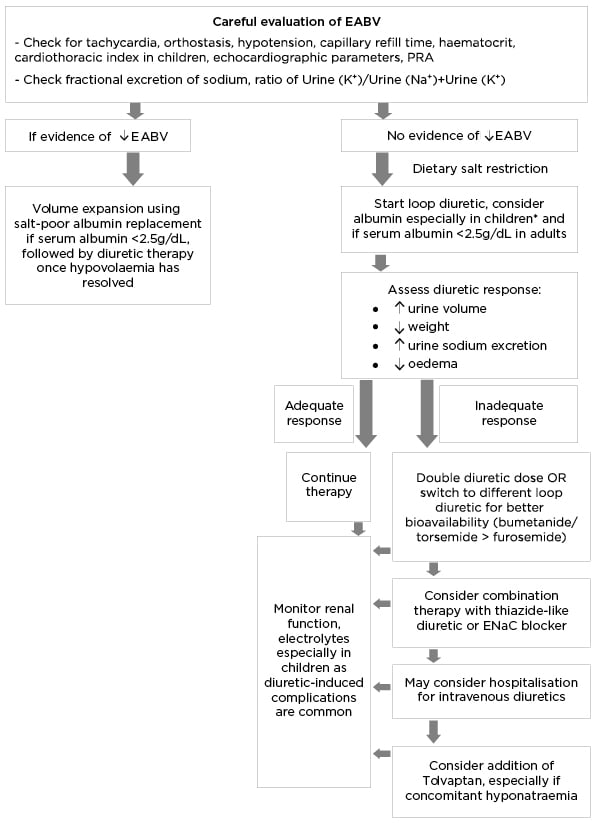
Figure 3: Proposed algorithm for the management of severe refractory oedema.
Fractional excretion of sodium <0.5%; Urine (K+)/Urine (Na+)+(K+) <0.6 indicate hypovolaemia.
*In paediatric NS, diuretics are typically indicated in steroid-resistant nephrotic syndrome or severe symptomatic oedema.
EABV: effective arterial blood volume; ENaC: epithelial sodium channel; NS: nephrotic syndrome; PRA: plasma renin activity.
Loop diuretics are the initial diuretic of choice in NS. They block Na+-K+2Cl– co-transporter in the thick ascending loop of Henle, where 25% of filtered Na+ is absorbed, making them potent diuretics. However, diuretic resistance is not uncommon. Intestinal oedema can decrease oral bioavailability of diuretics. Furosemide has lower oral bioavailability; therefore, bumetanide or torsemide might be preferable.44 Chronic loop diuretic use results in increased Na+ delivery to the distal nephron segments, resulting in significant Na+ absorption via ENaC. Distal nephron hypertrophy seen with prolonged diuretic therapy further enhances distal nephron Na+ absorption and failure of loop diuretic therapy.52 It is therefore recommended to attempt nephron blockade using a combination of diuretics acting at multiple nephron sites to maximise natriuresis and aquaresis. For example, a thiazide diuretic (inhibiting Na–Cl co-transporter in the distal convoluted tubule), direct ENaC inhibitors (amiloride), or mineralocorticoid antagonists (spironolactone) can be used in combination with a loop diuretic.
Amiloride may be particularly effective in NS due to the primary role of direct ENaC activation in oedema generation, as discussed above. Despite data regarding efficacy of amiloride in mouse models of NS and small human studies, it has not been studied in large clinical trials. In clinical practice, it is not used as a stand-alone diuretic therapy but is promising as an adjunct to loop diuretics.53,54 Other ENaC blockers such as triamterene have also been used successfully in combination with loop diuretics.44 Hyperkalaemia may be a limiting factor to the use of amiloride, especially in patients with diabetes and those with a low estimated glomerular filtration rate.
A small study (N=20) of patients with NS and refractory oedema demonstrated the benefit of acetazolamide and hydrochlorothiazide followed by furosemide therapy. This combination of diuretic therapy significantly increased 24-hour urine volume and decreased weight compared to the control group, who received furosemide and hydrochlorothiazide followed by furosemide alone. The study hypothesised that concomitant blockade of pendrin, a Cl––HCO3– exchanger in the intercalated cell of the collecting duct, by acetazolamide further potentiated the diuretic effects of thiazide and furosemide.55
It should be emphasised that diuretic use in NS requires close monitoring for acute kidney injury and electrolyte derangements, especially given variability in EABV, especially in children.
ALBUMIN COMBINED WITH DIURETIC THERAPY
There are two proposed benefits of using salt-poor albumin infusion with diuretic therapy in NS. Firstly, it may be required in oedematous patients with evidence of low EABV for the purposes of volume expansion prior to diuretic therapy. Secondly, it may be useful in combating diuretic resistance seen with severe hypoalbuminaemia. Loop diuretics are highly protein-bound and hypoalbuminaemia impairs delivery to their site of action, resulting in sub-optimal effectiveness. A small study (N=13) of NS with refractory oedema and diuretic resistance despite high doses of combination furosemide and spironolactone therapy showed that infusion of salt-poor albumin produced a prompt diuresis and weight loss.56 A cross-over study of 16 paediatric patients with refractory oedema, who were treated with furosemide and albumin versus diuretic alone, demonstrated greater weight loss and urine volume in those receiving albumin and furosemide, although there was no significant difference in Na+ excretion.57 Unfortunately, the data demonstrating benefit of albumin are weak and subsequent data have shown minimal efficacy.58-60 A recent Cochrane review determined that there was poor quality of published evidence to reach any definitive conclusions regarding the benefit of albumin in NS.61
Albumin may have an important role in paediatric NS and is often used in conjunction with diuretics in patients with severe oedema unresponsive to steroids. Excessive use can result in volume overload, and close monitoring for worsening hypertension and pulmonary oedema is recommended.51
AQUARETICS
Tolvaptan, a vasopressin receptor antagonist, reduces the density of luminal aquaporins in the principal cells of the collecting duct to increase water excretion (aquaresis) but not salt excretion. The use of tolvaptan has been described in patients with congestive heart failure and cirrhosis.62 More recently, case reports and small case series have shown benefit in combination with loop diuretics (increased urine output and decrease in oedema) in paediatric patients with NS, and may be especially useful in those with hyponatraemia.63-65 There are no clinical trials to confirm the benefit and there is risk of worsening hypernatraemia with its use. Other aquaretics, such as somatostatin analogues and urea channel inhibitors, may have a theoretical benefit, but are lacking data.45
CONCLUSION
Our understanding of the pathophysiology of oedema in NS has evolved over the last several years with newer insights into the roles of urinary plasmin, aldosterone independent ENaC activation, vascular hyperpermeability, paracrine factors, and interstitial inflammation in primary Na+ retention. While the underfill theory of hypoalbuminaemia-induced volume depletion and secondary RAAS activation applies to a subset of patients, especially children, the majority of the adult cohort is noted to be volume expanded in the setting of primary renal Na+ retention due to intra-renal factors. Management of oedema in patients with NS is a major challenge for clinicians due to significant clinical and biochemical heterogeneity, difficulty in assessing EABV, multifactorial aetiology, and issues around diuretic resistance. Well-designed clinical trials are needed to guide treatment, diuretic choice, and drug dosing, as well as to assess the benefit of albumin, direct ENaC blockers, and vasopressin antagonists in the treatment of oedema related to NS.