Abstract
The cure for Type 1 diabetes mellitus (T1DM) is likely to require an effective strategy for suppressing or evading the immune system. When considering curative treatments, it is almost inevitable to consider novel ways of inducing tolerogenicity to insulin-producing β cells. While the main mechanism of achieving tolerogenicity is restoring regulatory T cell (CD4+CD25+Fox3+) to effector T-cell (CD4+Fox3-) homeostasis, the means of achieving this are multifarious. The advent of a glucocorticoid-free immunosuppressive regimen was an early indication of how immunotherapeutics affect β-cell function. As newer biologics are developed, suppressing the immune system continues to become more specific and dynamic. An ever-evolving field of immunology has shifted the paradigm of how T1DM is understood, and the repurposing of T-cell-based biotechnology has the potential to change the way that it is treated. Regulatory T cells can be bioengineered to express T-cell receptors with affinity for peptide–human leukocyte antigen complexes that are frequently encountered in T1DM. Exosomes with embedded T-cell receptors can be isolated from regulatory T cells for use as an off-the-shelf therapy.
INTRODUCTION
While the precise aetiology and pathological mechanisms remain to be completely understood,1 Type 1 diabetes mellitus (T1DM) is a chronic metabolic disorder caused by the autoimmune destruction of insulin-producing β cells. The suboptimal release of insulin, at levels below the range required for metabolic homeostasis, is a consequence of the ample loss of β cells. T1DM is diagnosed by measurements of unusually high HbA1c and low levels of C-peptide, a byproduct of insulin production.2 The subcutaneous administration of exogenous insulin is currently the standard form of treatment. Along with the difficulties of precisely measuring and frequently administering insulin for appropriate conditions, the limitations of exogenous insulin administration include lifelong dependency, inadequate metabolic control, a moderate risk of inadvertently inducing severe hypoglycaemia, an undiminished risk of comorbidity, and reduced quality of life.3
The possibility for diagnostics to predict the risk of T1DM with high sensitivity and specificity, up to decades before its onset, seems to challenge the acceptance of autoimmune destruction of insulin-producing β cells as an inevitable fate of T1DM.4 The presence of genetic polymorphisms of human leukocyte antigen (HLA)-DQ and multiple autoantibodies that target islet self-antigens, which appear early in life, are highly predictive of T1DM.5 Autoantibodies target the islet cytoplasm (islet cell antibody [ICA]), native insulin (insulin autoantibody [IAA]), islet antigen-2 (IA-2), the 65-kDa isoform of glutamic acid decarboxylase (GAD65), and variants of zinc transporter 8 (ZnT8). Given that a small percentage of patients with T1DM demonstrate an absence of the aforementioned autoantibodies, non-HLA single nucleotide polymorphisms should be considered when determining combined risk.6,7 Before prevention becomes a standard form of treatment, diagnostics must be optimised for this purpose. To not be misled into treating false positives, it is critical to minimise the value of Type I errors and establish an acceptable threshold. Minimising Type II errors enables the opportunity to reduce the cumulative burden of morbidity in a population. Conceptually, prevention straightforwardly aims to preserve the interrelated mass and insulin-secretory function of β cells. There is early evidence that immunotherapy can delay the onset of the autoimmune destruction of β cells.8 Specific combinations of HLA haplotypes and autoantibodies are associated with increased risk for T1DM (Figure 1). Genetic screening for inherited HLA haplotypes identifies patients at risk for T1DM. In patients with at-risk HLA haplotypes, autoantibodies diagnostics inform its progression.
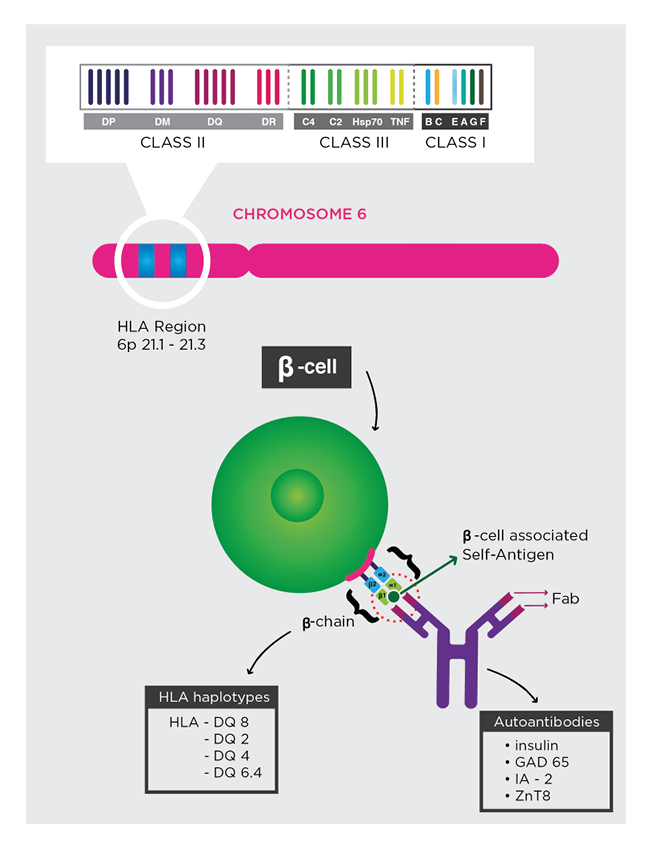
Figure 1: Genetic risk and autoantibody diagnostic.
Specific combinations of HLA haplotypes and autoantibodies are associated with increased risk for T1DM. Genetic screening for inherited HLA haplotypes identifies patients at risk for T1DM. In patients with at-risk HLA haplotypes, autoantibodies diagnostics inform its progression.
Fab: antigen-binding fragment; GAD65: the 65-kDa isoform of glutamic acid decarboxylase; IA-2: islet antigen-2; T1DM: Type 1 diabetes mellitus; ZnT8: zinc transporter 8.
While closed-loop insulin delivery using a control algorithm prototypes the sophisticated technology that mimics the glucose-responsive insulin secretion of β cells,9 β-cell replacement therapy in the form of islet transplantation is one of the few treatments demonstrating potential for insulin independence.10 The β cells present in an admixture of α cells, γ cells, δ cells, and ε cells within islets that have evolved closely together to release counter-regulatory hormones for metabolic homeostasis. The restoration of glycaemic control, a reduced risk of severe hypoglycaemia, the reversal of hypoglycaemia unawareness, and a reduced risk of comorbidity are the proposed clinically meaningful outcomes of islet transplantation. However, the duration of insulin independence is low and severe risks caused by complications or prescribed immunotherapeutics compromise its utility. Secondary immune deficiency is an unavoidable aspect of the intended effect and renal decline is not an uncommon side effect of the administration of nonspecific immunotherapeutics.11Therapies that aim to replace, preserve, or replicate β cells are limited by less-than-optimal immunotherapies. β-cell replacement therapy is, however, especially limited by a lack of β cell supply. Therefore, the regeneration of β cells from stem cells or even from the endogenous β-cell mass is an extraordinarily complex yet prominent area of research.12,13 The promise of treating autoimmunity early in life and/or empowering these other forms of curative treatments later in life is contingent on advancing approaches in immunotherapy.
IMMUNOSUPPRESSION IN THE EDMONTON PROTOCOL
Prior to the Edmonton protocol, anti-lymphocyte globulin and small molecules (cyclosporine, azathioprine, and glucocorticoids) were commonly used in a regimen as a means of nonspecifically attaining immunosuppression for islet transplantation.14 While glucocorticoids are widely used as an immunosuppressive steroid to treat autoimmunity,15 it is increasingly clear that glucocorticoids adversely stimulate gluconeogenesis in the liver and antagonise the insulin-mediated uptake of glucose.16 The induction of peripheral insulin resistance is counterproductive to the desired effect of islet transplantation. Enabled by an increase in newer immunosuppressive agents, the regimen included in the Edmonton protocol ventured with a glucocorticoid-free regimen consisting of sirolimus, tacrolimus, and daclizumab.
Sirolimus, also known as rapamycin, is a macrolide that binds to FKB12 and blocks the activation of the cell-cycle specific kinase TOR.17 Sirolimus indirectly inhibits the proliferation of T cells and B cells. Tacrolimus, discovered for its structural similarity to sirolimus, inhibits calcineurin with a much stronger potency compared to cyclosporine. A significantly lower dose of tacrolimus is therefore required to efficaciously induce immunosuppression and is considered the risk-averse alternative to cyclosporine.18 However, the risks associated with calcineurin inhibitors, including a decline in renal function and dialysis, are not sufficiently eliminated.19 Given that tacrolimus causes β-cell dysfunction and sirolimus biphasically induces insulin resistance,20 cotreatment with a GLP-1 agonist suggests that these counterproductive effects are reversible and can be prevented.21
Daclizumab, a monoclonal antibody that blocks the CD25 subunit of the IL-2 receptor, decreases IL-2 signalling at this high-affinity receptor. Due to the increased availability of IL-2, it inadvertently increases IL-2 signalling in cells expressing intermediate-affinity receptors.22 While daclizumab ameliorates autoimmunity with clinically meaningful effects, immune-mediated risks are concerns. Having received more scrutiny as severe, unintended effects are reported, including serious inflammatory disorders and death, its use is restricted and monitored.23 With the benefit to risk ratio as a priority, the immunotherapeutic regimen included in the Edmonton protocol is far from ideal.
BIOLOGICS AS OPTIONS FOR TARGETED IMMUNOSUPPRESSION
The immune system is regulated by cytokines. Upon binding to receptors on immune cells, cytokines turn on complex signalling pathways that activate key transcriptional factors. The transcriptional factors then promote the differentiation of naïve immune cells into specific lineages. In response to a large number of cytokines, naïve T cells differentiate into at least seven subtypes of helper (CD4+) T cells.24 While the subset of helper T cells is in an adjustable equilibrium, a disequilibrated regulatory T-cell (Treg) to Th17 cell balance leads to autoimmunity.25 The TGFβ/IL-6 and IL-2 cytokine axis regulates the differentiation of naïve T cells into either the Treg cell or Th17 cell lineage.26 Via the TGFβ and IL-6-mediated activation of transcription factors STAT3 and RORγt, Th17 cells are involved in the autoimmune destruction of β cells.27 Whereas Treg cells inhibit autoreactive CD8+ T cells,28 Th17 cells activate autoreactive CD8+ T cells. Autoreactive T cells are known to either escape clonal deletion or differentiation into the thymic Treg cell lineage early in life and enter the peripheral lymph nodes of the pancreas.29 Biologics are the cytokines or antibodies that are manufactured for therapeutic purposes, such as manipulating the cytokine axis to re-enact an equilibrated Treg–Th17 cell balance.
DNA recombinant technology has enabled the engineering of recombinant cytokines and antibodies with specific targets. Subsequent to antigen presentation, IL-2 triggers the expansion of CD25+ Treg cells.30 Given that CD25+ Treg cells express high-affinity IL-2 receptors, a low dosage of IL-2 is sufficient to trigger the expansion of CD25+ T cells.31 A low dosage of IL-2 may be a strategy to mimic the Treg–Th17 cell homeostasis. However, other immune cell types also express high-affinity IL-2 receptors. Therefore, it is possible for a long-term low dosage of IL-2 to backfire. Monoclonal antibodies JES6-1 and F5111.2 are strategically attached to IL-2 to stabilise a conformational change that increases its selectivity for the high-affinity receptors on CD25+ Treg cells.32 This strategy averts off-target effects that are likely to backfire, and can guide the development of antibodies for immunomodulatory cytokines with similar pharmacodynamics. Humanised versions of JES6-1 and F5111.2 are in development.
Whereas an increase in clinical trials indicates a renewed interest in cytokines,33 antibodies that target cytokines or the corresponding receptors are more commonly used. Monoclonal antibodies, such as adalimumab and etanercept, bind to TNFα with a higher affinity compared to its TNFα receptors.34 The binding of adalimumab to TNFα induces a conformational change that trimerises TNFα receptors on Tregs and triggers its expansion.35 More commonly, antibodies are used to suppress Th17 signalling. Brodalumab is a monoclonal antibody that binds to the IL-17 receptor and is approved for certain autoimmune diseases.36While preclinical studies suggest that Th17 cells are also involved in T1DM,37 clinical trials are required to reveal whether antibodies that suppress Th17 signalling have therapeutic effects in T1DM. Teplizumab, an Fc-receptor non-binding, anti-CD3 monoclonal antibody, obstructs the transmembrane assembly of CD3 subunits within the T-cell receptor (TCR).38 This prevents signalling downstream of the TCR and therefore mimics effector T-cell exhaustion.39 A randomised, double-blind Phase II trial demonstrated that a single 14-day course of teplizumab delayed the onset of T1DM by 24.4 months compared to a placebo-treated group.40 The U.S. Food and Drug Administration (FDA) has granted teplizumab a breakthrough therapy designation to efficiently expedite the process of determining whether there is more evidence to support its approval.41
Given a lack of randomised controlled trials comparing the efficacies of different biologics for T1DM, a post hoc study was used for insight into how the outcomes of islet transplantation are related to the type of biologics used.42 The biologics in this study included an Fc-receptor non-binding, anti-CD3, antithymocyte globulin (ATG) or alemtuzumab alone; ATG or alemtuzumab with TNFα inhibitors; and anti-CD25. The duration of insulin independence provided by transplanted islets was increased when Fc-receptor non-binding, anti-CD3 and ATG, or alemtuzumab with TNFα inhibitors are used. Given that CD4+Fox3- cells express higher levels of CD3 compared to CD4+Fox3+ cells, the anti-CD3 is expected to target the effector CD4+ T cells while mostly sparing Treg cells.43 ATG, a polyclonal antibody that suppresses lymphocytes and other immune cell types through diverse mechanistic pathways to prevent acute rejection, is shown to induce the expansion of Treg cells ex vivo.44 Alemtuzumab, an anti-CD52 monoclonal antibody, similarly depletes lymphocytes.45 A single course of low-dose ATG delays the decline of C-peptide levels at 1 year after infusion.46 Because ATG and antibodies against TNFα antibodies have demonstrated potential to expand Treg cells, there is consistency with inducing tolerogenicity to transplanted β cells via restoring Treg to effector T-cell homeostasis.
Many of the biologics used in cancer immunotherapy are repurposed for autoimmunity. Among these, biologics that target costimulation and co-inhibition are well-known (Figure 2).47 When an effector T cell recognises an antigen presented on either major histocompatibility complex (MHC) I or II, the B7 family binds to either costimulatory CD28 or co-inhibitory CTLA-4.48 Binding of the B7 ligands to CD28 is necessary for activation of effector T-cell function. CTLA-4 competes with CD28 for binding to B7 ligands to inhibit this activation. Given that CTLA-4 binds to B7 ligands with a higher affinity, recombinant CTLA-4 fused to Ig, such as abatacept and belatacept, attempt to exploit co-inhibition. CTLA-4 is fused to Ig to increase its half-life. Clinical trials prove that abatacept delays the decline of C-peptide levels in recent-onset T1DM.49 In principle, anti-CD28 should block costimulation; instead, anti-CD28 facilitates the homodimerisation of CD28 and serves as an agonist.50 While used as an agonist for cancer immunotherapy, the anti-CD28-mediated cytokine storm serves as a reminder of the risks involved when using biologics to manipulate the immune system.51
While preclinical and clinical studies suggest that these biologics have potential to ameliorate autoimmunity, none have been approved for T1DM. However, teplizumab is the first to receive a breakthrough therapy designation by the FDA for T1DM.
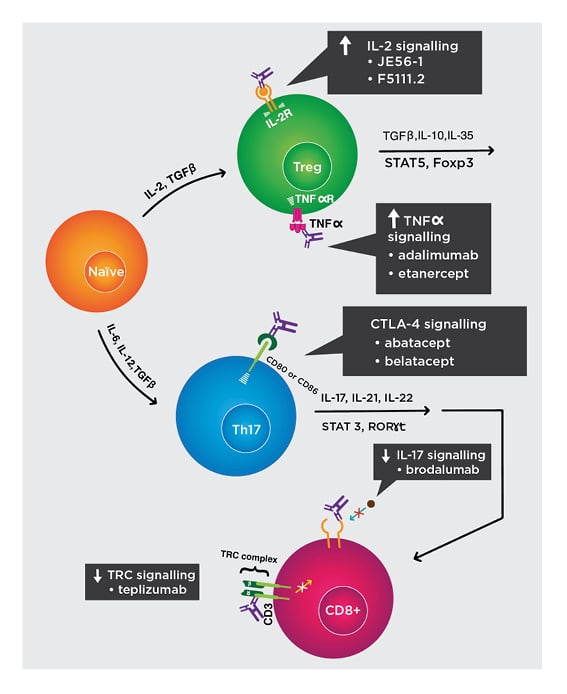
Figure 2: Biologics to restore regulatory T cell to effector T-cell homeostasis.
Mechanisms of biologic activity:
- Increase IL-2 signalling. JES58-1 and F5111.2 preferentially bind to IL-2 receptors on Treg cells and induce its expansion. Humanised versions are in development.
- Increase TNFα signalling. Adalimumab and etanercept trimerise TNFα receptors on Treg cells and induce its expansion.
- Increase CTLA-4 signalling. Recombinant CTLA-4 fused to Ig, such as abatacept and belatacept, exploit co-inhibition and divert away from CD28 co-activation.
- Decrease IL-17 signalling. Brodalumab binds to IL-17 receptors to suppress downstream signalling that activate autoreactive CD8+ T cells.
- Decrease TCR signalling. Teplizumab binds to the CD3 subunits of TCR and prevents downstream signalling that activate T cells.
IL-2R: IL-2 receptor; TCR: T-cell receptor; TNFαR: TNFα receptor; Treg: regulatory T cell.
Adapted from Raffin et al.47 The concept and content of the figure is the authors’ own.
THE POTENTIAL OF CELL-BASED IMMUNOTHERAPY
Cells integrate environmental signals to execute complex, regulated behaviour. Infusing cells with immunosuppressive therapeutic behaviours, such CD4+CD25+Fox3+ Treg cells and mesenchymal stem cells (MSC), is potentially a more dynamic strategy for restoring tolerance to β cells.52 The loss of function of Fox3+, an important transcription factor involved in the differentiation of naïve T cells into Treg cells, causes immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome.53 In an adolescent patient with late-onset IPEX syndrome with T1DM, allogeneic haematopoietic stem cell therapy (aHSCT) delays the decline of C-peptide levels for 15 months.54 aHSCT reconstitutes an equilibrated population of functional Treg cells and other immune cells from a donor. Given the significant morbidity caused by graft-versus-host disease (GVHD), the clinically meaningful outcomes derived from aHSCT outweigh the morbidity of GVHD only for T1DM patients with underlying IPEX syndrome. The adoptive cell transfer of autologous Treg cells may be the safer counterpart to aHSCT for the majority of T1DM patients who do not have underlying IPEX syndrome. For the adoptive cell transfer of autologous Treg cells, extirpating the recipient’s immune system is unnecessary and morbidity from GVHD is not a concern. Several good manufacturing practice-compliant protocols for the isolation of autologous Treg cells from peripheral blood have been established.55 Isolation of a low quantity of Treg cells is sufficient given that small molecules and biologics can be used to induce the ex vivo expansion of Treg cells with high purity.56
The adoptive cell transfer of autologous Treg cells induces tolerogenicity57 and clinical trials are underway to determine whether it can efficaciously ameliorate autoimmunity. Treg cells are more potent when genetically engineered to express chimeric antigen receptors (CAR) or TCR that bind preferentially to peptide–HLA complexes.58 Whereas CAR bind to peptide–HLA complexes at higher affinities, binding to higher quantities of antigen is a requisite for sufficient activation of signalling downstream of CAR.47 TCR bind at lower affinities yet are activated in the presence of lower quantities of antigen. Therefore, TCR Treg cells are appropriate to autoimmunity considering that the CD4+ T cells with the peptide–HLA complexes of interest are infinitesimal. With CRISPR/Cas 9, the endogenous alleles for the α and β chains of TCR can be precisely cut and replaced with the alleles for the α and β subunits of TCR engineered to target the peptide–HLA complexes of interest.59 The isolation of autoreactive T cells and overexpression of Foxp3 is another approach but less common because of issues with instability.60 Since Foxp3 is critical for maintaining the functionality of Treg cells, targeting epigenetic regulators and post-transcriptional modifiers are opportunities to enhance stability.61
One of the main safety concerns for Treg cells is the ability to lose Foxp3 expression and acquire an effector T-cell phenotype. Engineering suicide signalling pathways that can be easily triggered by small molecules, in the event that Treg cells become unstable, is a strategy to prevent from paradoxically exacerbating autoimmunity. The final TCR Treg cell product can be expanded using IL-2 and CD28 superagonists.62 Since manufacturing TCR Treg cells is a labour-intensive and time-restraining process, it is challenging to have this therapy readily available. Determining which peptide–HLA complexes are distributed at higher frequencies among the genetic pool of a representative sample of T1DM patients can inform which TCR Treg cells can be manufactured for off-the-shelf use. Given that the TCR Treg cells are not recognised by the recipient’s host system, novel approaches are needed.
Multipotent MSC are therapeutic cells that have been extensively studied for diverse purposes. Via paracrine secretion, MSC release TGFβ, prostaglandin E2, hepatocyte growth factor, indoleamine-pyrrole 2,3-dioxygenase (IDO), nitric oxide, IL-2, IL-4, IL-10, and galectin-1.63 In the presence of the TGFβ and IL-2, naïve T cells are known to differentiate and expand into CD25+Fox3+ Treg cells. With the exception of hepatocyte growth factor, the released molecules mediate immunosuppression. By catalysing the rate-limiting step of tryptophan metabolism, IDO renders effector T cells and dendritic cells ineffective.64 While nitric oxide is an immunomodulator, its activity is concentration-dependent yet not uniform.65 A dose- and frequency-dependent association between the infusion of MSC and the preservation of insulin secretion suggest causality.66 When infused, MSC reduced exogenous insulin requirement by one-half for 2 years and curbed HbA1c levels for 3 years.67 While MSC are the most clinically studied cell-based therapy, inconsistent results have clouded therapeutic efficacy and have indefinitely delayed FDA approval.68 Inconsistencies are partially attributable to differences in cell source and culturing practices.69 MSC are determined to be moderately safe, albeit with concerns for tumourigenicity and the ability to become trapped in the lung microvasculature.
EXOSOMES AT THE FRONTIER AS AN OFF-THE-SHELF IMMUNOTHERAPY
Exosomes are released via autocrine, paracrine, or endocrine signalling by most cells for intracellular or intercellular communication with neighbouring and distant cells. Minute (30–150 nm) spherical sacs of phospholipid bilayer, exosomes enclose a cargo of proteins, mRNA, and microRNA (miRNA).70 Exosomes are isolated from the interstitial fluids or specific cell types for diagnostic or therapeutic purposes. Whereas analysing changes in the cargo of plasma-derived exosomes can potentially track the progression of T1DM,71exosomes that are isolated from specific cell types are a potentially versatile therapeutic tool.
MSC-derived exosomes enclose IL-2, IL-4, IL-10, TGFβ1, IDO, proteins, and miRNA that are suggested to regulate the expression of IL-6, IL-17AF, IL-12p70, and IL-22.72,73 MSC-derived exosomes are preferred over MSC because of a lack of tumourigenicity, lower risk of becoming trapped in the lung microvasculature, ability to evade immune recognition, and modifiability of the cargo via transfection with therapeutic nucleic acids.74 While preclinical studies suggest that the protective effects of MSC-derived exosomes are due to suppressed differentiation of naïve T cells into the Th17 cell lineage, the first clinical trial to investigate the potential of MSC-derived exosomes to ameliorate T1DM has not yet published results.75 Given that exosomes can be derived from MSC of different origins and manufacturing practices, it is necessary to ensure that protocols comply with good manufacturing practice and establish in vitro assays that verify potency.
Whereas CD8+ T cells perform cytotoxic activity in a contact-dependent manner, CD4+ T cells mostly perform via paracrine signalling. Treg cells are a rich source of exosomes that contain immunosuppressive proteins, mRNA, and miRNA. Developed via transfection with the dominant negative form of IKK2, Fox3+CD25- Treg cells secrete exosomes with a unique set of miRNA and isoform nitric oxide synthase mRNA.76 When engulfed by target T cells, the miRNA and isoform nitric oxide synthase from these exosomes inhibit the transcription of cell-cycle proteins and induce apoptosis. Whereas T cells are targeted by direct exposure to exosomes in ex vivo assays, a strategy that facilitates the in vivo engulfment of exosomes by autoreactive T cells is needed. Exosomes are formed by inward buddings of endosomal vesicles derived from the plasma membrane.77 Isolating exosomes from TCR Treg cells is a potential strategy, because engineered TCR are embedded within the membrane of the exosomes. To increase the output of the exosomes with embedded TCR per cell, expression of the engineered TCR needs to be enhanced (Figure 3).47 While it is unclear whether binding of the TCR to peptide–HLA complexes can be exploited to induce uptake of exosomes, the exosomes are steered toward the autoreactive T cells and accumulate in its vicinity. Clarifying the cellular mechanisms that regulate delivery of exosomes into specific targets can reveal other strategies to improve its uptake. These mechanisms, such as overexpressing key receptors for receptor-mediated endocytosis,78 can be exploited to induce uptake of exosomes. Exosomes with embedded TCR that recognise the peptide–HLA complexes most frequently encountered in T1DM can be manufactured for off-the-shelf therapeutic use. Exosomes are generally safer than cell-based therapies because of a lack of tumorigenicity and a lower risk of becoming trapped in lung microvasculature. However, there is a chance for exosomes to be cleared by the immune system if integral proteins embedded in the membrane are immunogenic. By knowing the sources of immunogenicity, strategies such as genetic editing can be used to overcome this issue.
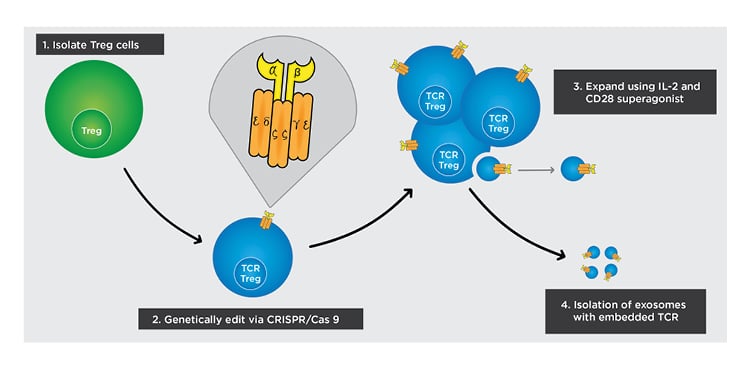
Figure 3: Cell-based approach to inducing tolerogenicity.
The following steps outline how off-the-self exosomes can be manufactured from TCR Treg cells in a simplified diagram.
1) Isolate Treg cells from peripheral blood.
2) Deliver genes for the α and β chains of the engineered TCR into Treg cells and use CRISPR/Cas 9 to replace endogenous genes with delivered genes.
3) Isolate the engineered TCR Treg cells from non-engineered Treg cells and expand using an IL-2 and CD28 superagonist.
4) After culturing TCR Treg and increasing its expression of TCR, isolate exosomes with TCR embedded in the membrane.
TCR: T-cell receptor; Treg: regulatory T cell.
Adapted from Raffin et al.47 The concept and content of the figure is the authors’ own.
CONCLUSION
While insulin delivery systems mimic the glucose-responsive behaviour of β cells, the insulin-release kinetics are inferior to mature β cells. Mature β cells evolved complex mechanistic signalling pathways to regulate glucose-responsive insulin secretion. Preserving, regenerating, and replacing β cells are treatments with the potential for insulin independence. However, these treatments are limited by inadequate and risk-prone immunosuppression. Given that the cure for T1DM is likely to require an effective strategy for suppressing or evading the immune system, advanced immunotherapies are needed. Whereas biocompatible encapsulation is one of the undiscussed approaches that seek to evade immune recognition, other immunotherapies seek to induce tolerogenicity to β cells. Biologics, immunomodulatory cells, and exosomes are exploited to restore Treg cell to effector T-cell homeostasis and thereby induce tolerogenicity to β cells. Exosomes are at the frontier with the potential to become an off-the-shelf therapeutic tool. Having TCR embedded within the exosome membrane that bind preferentially to peptide–HLA complexes, exosomes isolated from TCR Treg cells are a promising immunotherapy. With selectivity for autoreactive T cells, these exosomes are more potent and keep other immune defenses intact. At time of writing, this is a novel concept that has not yet mutated into invention.