Abstract
The role of different lipid species such as free fatty acids and sphingolipids in non-alcoholic fatty liver disease (NAFLD) has been extensively studied during the last decade. In addition, free cholesterol accumulation in hepatocytes plays a crucial role in the transition from steatosis to steatohepatitis. However, the contribution of these lipids to NAFLD pathology is often evaluated individually. This review attempts to enclose the main metabolic and signalling connections between lipotoxic lipid species, and how their homeostasis is disrupted in NAFLD.
INTRODUCTION
In 1953, Hokin and Hokin discovered that some lipid species located in the cell membrane were able to participate in cell signalling.1 Since then, the role of lipids in different pathologies has become an intense area of research; despite this, the pathogenesis of non-alcoholic fatty liver disease (NAFLD) is still poorly understood. The most prevalent hypothesis, proposed in 1998,2 postulates the convergence of two hits, the first consisting of the accumulation of lipid droplets or steatosis, followed by the onset of the second hit, which induces inflammation and tissue damage, defined as steatohepatitis. This hypothesis implies that steatosis is a sensitisation factor for the reactions of the second hit. Due to this preconception, many in vivo dietary studies have been performed with unspecific high fat diets, sometimes without a lipid composition disclosure and often named as ‘Western diets’.3,4 While this is a useful approach to achieving some clinical features found in patients, such as steatosis, it hampers elucidation of the key detrimental lipid species that are participating in stress, inflammation, and apoptosis signalling pathways. There is compelling evidence that hepatic triglycerides (TG) are not the effectors of cellular toxicity in NAFLD, but rather a defence mechanism to avoid free fatty acid (FFA) accumulation, which can trigger cell death pathways.5 Likewise, many reports suggest an important lipotoxic role of cholesterol and sphingolipid species.6,7
Fatty liver disease therefore has a complex pathology, which comprises many concomitant cell alterations.8 In patients, plasma serum and liver biopsies show different lipid signatures for each stage of the pathology.9-12 This connection between lipid composition and disease severity underscores how important it is to further characterise the contribution of these molecules to NAFLD pathology and how they interact with each other in order to advance treatment and prevention strategies.
The purpose of this review is to describe the lipid metabolic changes induced by the main lipotoxic lipids in NAFLD.
FATTY ACIDS
Fatty acids (FA) are carboxylic acids with a non-ramified aliphatic chain of different length. In mammals, FA usually contain up to 28 carbons in even numbers, but the most abundant length in biological tissues ranges from 14–20. In addition, the presence of one or more double bonds between carbons further increases the diversity of these molecules. Saturated FA (SFA), with no double bonds, account for 30–40% of the FA present in animal tissues, in order of abundance: palmitic acid (PA) (15–25%), stearic acid (10–20%), myristic acid (MA) (0.5–1%), and lauric acid (<0.5%).13
The role of medium-long-chain FA in NAFLD has been more extensively studied than shorter chain FA (<16 carbons), probably because of the faster catabolism of the latter and therefore lower evidence of toxic effects in the cell.14 Nowadays, it has been demonstrated that FFA species can have opposite roles in NAFLD, particularly when it comes to comparing SFA with unsaturated ones. Buettner et al.15 observed that high fat diets with different fatty acid compositions triggered different outcomes: a high intake in polyunsaturated FA reduced expression of lipogenic genes regulated by sterol regulatory element-binding protein 1 (SREBP-1c) and increased peroxisome proliferator activated receptor (PPAR)-α-regulated lipolytic genes, whereas diets rich in SFA and monounsaturated FA induced hepatic steatosis. PA is the source of FA and ceramide synthesis in the endoplasmic reticulum (ER) of the cell and is also the most abundant FA in Western diets. However, an intracellular increase in non-esterified PA can be fatal, since this molecule induces mitochondrial dysfunction,16 lysosomal permeabilisation,17 ER stress, and autophagy alteration.18 Similar lipotoxic effects have been observed with other SFA such as stearic acid.19,20 On the other hand, many in vitro studies report that unsaturated fats like oleic acid could protect cells against PA lipotoxicity.21-23
The line between ‘good’ and ‘bad’ blurs when considering insulin resistance in the literature. Buettner et al.15 found that an olive oil enriched diet led to insulin resistance in rats. Conversely, it has been shown that the polyunsaturated FA oleic acid, the main olive oil component, has the ability to induce PPAR-α thereby promoting FFA oxidation, safe storage into TG, and conferring protection against insulin resistance.24 Of note, studies that report the participation of unsaturated fats in insulin resistance are often performed with lipid mixtures,25 making it difficult to exclude a possible intervention of other lipid species in the pathogenic mechanism.
FATTY ACIDS AND SPHINGOLIPIDS
PA is the precursor of ceramide, the simplest sphingolipid, which belongs to a family of lipids present in biological membranes and segregated in particular domains, where they participate in different signalling events. Sphingolipid synthesis occurs in the ER and begins with the condensation of the amino acid L-serine with PA to form 3-ketosphinganine. This first reaction of the pathway is achieved by the enzyme serine palmitoyltransferase and is the limiting step. Afterwards, 3-ketosphinganine reductase reduces 3-ketosphinganine to sphinganine followed by action of dihydroceramide synthase, which adds a fatty acid to sphinganine to form dihydroceramide. The six dihydroceramide synthase isoforms described so far exhibit specificity towards different FA, giving rise to heterogeneous ceramide species.26 Finally, dihydroceramide desaturase (DES) creates a double trans 4,5 bond thereby converting dihydroceramide into ceramide. Interestingly, Beauchamp et al.27 found that DES can be modified by an irreversible lipidation in its N-terminal residue with MA through a process called myristoylation, which is carried out by the enzyme N-myristoyltransferase. Myristoylation increases the activity of DES implying that MA, a 14 carbon saturated FA present in mammalian milk, can potentiate the synthesis of ceramide. Once synthesised, ceramide traffics towards the Golgi apparatus, where it is modified to generate more complex sphingolipids: ceramide trafficking, by the ceramide transfer protein to the Golgi, is used for sphingomyelin synthesis by the enzyme sphingomyelin synthase through the addition of a phosphocholine or phosphoethanolamine group in the first carbon of ceramide with concomitant generation of diacylglycerol.28 Alternatively, ceramide can also travel to the Golgi apparatus by vesicular transport to be converted into glucosylceramide upon the addition of glucose by the enzyme glucosylceramide synthase. The addition of sialic acids to glucosylceramide generates complex sphingolipids and gangliosides. Besides the aforementioned de novo synthesis, ceramide can also be generated from acid (ASM) or neutral (NSM) sphingomyelinases and from the internalisation of membrane sphingolipids through endocytic pathways to the lysosomes, where they are hydrolysed by sphingomyelinases or glucosidases to produce ceramide, a process known as the salvage pathway. Many routes therefore converge into the generation of ceramide. Importantly, ceramide and other sphingolipid species are bioactive lipids that contribute to a myriad of cellular processes; the alteration of their metabolic homeostasis is a potential therapeutic target, based on modulating their levels towards a desired functional outcome.29
In a fatty liver scenario, the increase in PA due to its higher dietary intake and liver import fuels de novo ceramide synthesis. Moreover, PA and other SFA can act as toll-like receptor 4 agonists,30 a signalling pathway shown to induce ceramide synthetic enzymes in macrophages,31 but whether this occurs in hepatocytes is still under debate.32,33 PA could also indirectly trigger ceramide synthesis through its lipotoxic effects. For example, reactive oxygen species or cytokines like tumour necrosis factor alpha (TNF-α) lead to a rapid accumulation of ceramide through ASM.34 This event is of particular relevance in lipid rafts, membrane microdomains with a high concentration of sphingolipids, cholesterol, and signalling molecules. The produced ceramide is capable of spontaneously self-aggregating and reorganising lipid rafts, thereby fusing them and creating large signalling platforms. Consequently, ceramide plays a crucial role in cellular signalling by clustering receptors together.35
Similarly to PA, ceramide accumulation triggers lipotoxic pathways like inflammation, ER stress, mitochondrial dysfunction and permeabilisation, autophagy alteration, and lysosomal cathepsin D activation, as reviewed in detail elsewhere.25,36-38 Some of these pathways can in turn induce ceramide and toxic derivatives like gangliosides, creating a vicious cycle. On the other hand, sphingosine-1-phosphate or ceramide-1-phosphate have been reported to be anti-apoptotic and mitogenic.29 Recently, an effort has been made to decipher the role of different ceramide species in cellular signalling. Ceramide C16:0 is toxic for macrophages39 but the contrary has been observed in human head and neck squamous cell carcinomas, where C16:0 was anti-apoptotic and C18:0 pro-apoptotic.40 Therefore, the role of ceramide species seems to be specific to each cell type and further investigation is required to characterise their cellular effects.
It is important to note that there is an overlap of the targeted organelles and lipotoxic outcomes reported with PA and its product ceramide in the literature. Since PA can be converted into ceramide or trigger ceramide synthesis by other means, particular care is suggested before attributing lipotoxic roles to PA. In this regard, we have recently determined that ER stress is aggravated by de novo synthesised ceramide after PA treatment in primary hepatocytes.41 Importantly, we have also validated in vivo the potential toxic role of MA when ingested in combination with PA through a sustained ceramide production, causing serious disruption of lipid metabolism homeostasis, ER stress, inflammation, and steatohepatitis.
FATTY ACIDS AND CHOLESTEROL
Cholesterol is a crucial sterol for animal cell membranes. The molecule intercalates between phospholipids, positioning the hydroxyl group near the polar and the steroid rings in the apolar zones. This interaction immobilises and packs the membrane, diminishing its fluidity and permeability, but at the same time protects the membrane against phase transition due to a higher distance between aliphatic chains. Besides its structural function, cholesterol is highly abundant in lipid rafts, where it participates in the formation of caveolae, and is the precursor of steroid hormones, biliary acids, and vitamin D.
Synthesis of cholesterol is regulated by its availability. The main negative feedback mechanism is the inhibition of the sterol regulatory element-binding protein 2 (SREBP-2), a transcription factor that activates cholesterol biosynthetic and uptake enzymes like 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and low-density lipoproteins (LDL), respectively. Similarly, cholesterol can also directly bind to HMG-CoA reductase to induce its degradation by the proteasome.42 A high dietary intake of cholesterol therefore reduces its endogenous synthesis. This regulation might be compromised in case of a concomitant increase of other lipid types, mainly TG, which provide a source of FA. FA β-oxidation would produce acetyl-CoA, required for the first step of de novo cholesterol synthesis through the mevalonate pathway. However, acetyl-CoA availability is controlled by the different subcellular localisation of these metabolic pathways, meaning that previous export from the mitochondria to the cytosol, through the citrate transporter, is required.43
Cholesterol homeostasis can also be regulated by the nuclear receptors liver X receptor (LXR) and farnesoid X receptor (FXR). Oxysterols activate LXR, inducing the transcription of genes involved in cholesterol transport and clearance, thereby making it a potential therapeutic target for atherosclerosis.44 On the other hand, LXR activation also induces lipogenic transcription factors like SREBP-1c, meaning that cholesterol could indirectly influence steatosis. In the context of fatty liver disease, LXR inhibition instead of activation may therefore be more beneficial.45
Cholesterol catabolism into bile acids begins with its conversion to 7-α-hydroxycholesterol by cholesterol 7-α-monooxygenase (CYP7A1), the rate-limiting step of the pathway.46 Bile acids bind FXR, which represses CYP7A1, acting as a negative feedback loop. Besides its role in maintaining the balance between cholesterol and bile acids, FXR also induces lipolysis by activating PPAR-α and repressing SREBP-1c.47 Accordingly, FXR deficiency in mice results in a hepatic and serum increase of cholesterol and TG,48 which are NAFLD hallmarks. FXR activation by natural and synthetic agonists is thus currently regarded as one of the main therapeutic strategies for NAFLD.49 However, clinical studies show that treatment with FXR agonists has some undesired side effects such as pruritus or increased LDL cholesterol,50 so further research is required to develop FXR modulators with a higher specificity for lipolytic activity.
The increase in lipotoxic FFA, such as PA, in the fatty liver promotes ER stress, which has been shown to activate SREBP-2.51 In relation to this, we have recently observed that mice that are fed diets with PA or PA plus MA have ER stress, higher HMG-CoA reductase gene expression, and hepatic cholesterol.41 Curiously, a high intake in MA, which is not regarded as lipotoxic per se, has been found to affect serum lipoproteins and hepatic cholesterol levels.52 The authors attributed this change to a negative correlation of MA with the scavenger receptor Class B Type I, a high density lipoprotein receptor, and cholesterol catabolic enzymes, but they did not analyse ER stress markers. Moreover, since the authors used a mixture of different fats, the participation of other lipid types in this outcome cannot be discarded.
Conversely, an increase in ER membrane cholesterol-loading can perturb the organelle calcium stores, thereby triggering ER stress and engaging in a vicious cycle.53 The unfolded protein response induces lipogenic genes expression, enhancing the accumulation of FA, lipotoxicity, and synthesis of other lipotoxic lipids like ceramides. Our group has also reported that free cholesterol accumulation can affect other organelles like mitochondria, by depleting mitochondrial glutathione and sensitising hepatocytes to inflammatory cytokines54 and lysosomes, by impairing mitophagy and the subsequent removal of damaged mitochondria.55
CHOLESTEROL AND SPHINGOLIPIDS
The relationship between cholesterol and sphingolipids has been less directly addressed, but these two lipids seem to be tightly regulated. Cholesterol preferentially binds to sphingomyelin in lipid membranes and concomitant changes in their levels have been reported in several studies, as reviewed in detail by Ridgway56 and briefly synthesised in the following lines. An obvious link between cholesterol and sphingomyelin is their presence and crucial role in lipid rafts, suggesting that a proper ratio is necessary for the functioning of these particular membrane regions. In fact, a sphingomyelin decrease in the plasma membrane by exogenous sphingomyelinase treatment downregulates SREBP-2, and induces cholesterol esterification and its retrograde transport towards the ER. However, it is unclear if the local ceramide formation by this sphingomyelin hydrolysis participates in the process. In contrast, a decrease in cholesterol may not mean a decrease in sphingomyelin. This might be due to the need for sphingomyelin to continue other important functions for the cell and to maintain the membrane structure.
An imbalance of these lipids is seen in a number of pathologies. Niemann–Pick diseases are inherited lysosomal storage disorders with severe metabolic and in some cases neurological consequences. Acid sphingomyelinase deficiency and resultant sphingomyelin accumulation in lysosomes is the cause of Niemann–Pick Type A and B, whereas Type C (NPC) is caused by NPC 1 or 2 protein deficiency, involved in cholesterol efflux from lysosomes. A hallmark of all Niemann–Pick variants is fatty liver disease, with accumulation of sphingomyelin, cholesterol, and other lipid types in lysosomal compartments and other cell locations.57,58 Due to the close relationship between cholesterol and sphingomyelin and because these pathologies share common traits, treatment of patients with sphingomyelinases could be an effective therapy regardless of their genetic defect. In fact, NPC patients also have a low ASM activity due to secondary sphingomyelin accumulation, and treatment with sphingomyelinases has proven effective against cholesterol loading in the lysosomes of NPC cells.59
Finally, and similarly to SFA, other sphingolipids like ceramide and gangliosides could induce ER stress,60 thereby activating SREBP-2 and cholesterol synthesis, as we have recently observed in vivo.41 Cholesterol levels can therefore be influenced by sphingolipids or vice versa and further research will be required to discern their respective lipotoxic roles in NAFLD.
LIPID HOMEOSTASIS AND GENETIC POLYMORPHISMS
Comparative studies of candidate genes in NAFLD patients have found some polymorphisms associated with an altered lipid metabolism. For example, a single nucleotide polymorphism (SNP) in the microsomal triglyceride transfer protein (MTTP) was identified in its promoter region.61 MTTP is involved in very low density lipoprotein (VLDL) synthesis and export, but patients with the SNP variant are predisposed to liver and serum TG accumulation due to reduced MTTP protein expression. However, experimental design with larger cohorts might be required to confirm findings in comparative studies, since other works often do not report the same correlations.62
Genome-wide association studies have also identified different SNPs associated with NAFLD, as reviewed in detail by Anstee and Day.63 Among them, the SNP rs738409 (Ile148Met) in the patatin-like phospholipase domain-containing 3 (PNPLA3) gene is currently the most strongly associated with TG accumulation and NAFLD biochemical markers. PNPLA3 is a triacylglycerol lipase, but curiously the SNP presence is not only related to steatosis but also to steatohepatitis and fibrosis.64 Further research is therefore needed to fully elucidate the mechanisms by which this allelic variant promotes such pathogenic outcomes.
CONCLUSION
Lipids play a crucial role in cell signalling pathways and metabolism. The study of individual lipid species is a powerful tool to look for possible therapeutic strategies, but it is also important to understand the way these molecules’ metabolic and signalling pathways are interconnected. Figure 1 summarises how the main lipotoxic species can affect each other in the context of fatty liver and Figure 2 shows the main pathways that disrupt lipid homeostasis. NAFLD research has evolved towards a hunt for detrimental lipids. Considering the interconnectivity summarised in this review, in vitro and in vivo studies should systematically perform a detailed quantification of lipid species and metabolic gene expression, which should be regarded as necessary as assessing transaminases in serum. For that purpose, lipidomics have become crucial in identifying changes in a myriad of these molecules with high sensitivity,65 thereby providing the basis to look for molecular pathogenic mechanisms.
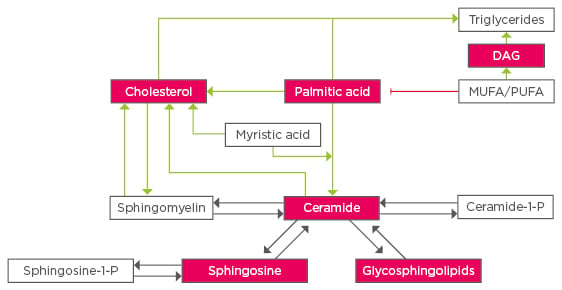
Figure 1: Crosstalk between lipid species in fatty liver disease.
Lipotoxic lipid species are highlighted in red, dark arrows represent enzymatic reactions, green and red arrows refer to changes through stress or metabolic pathways.
MUFA: monounsaturated fatty acids; PUFA: polyunsaturated fatty acids; DAG: diacylgylcerol.
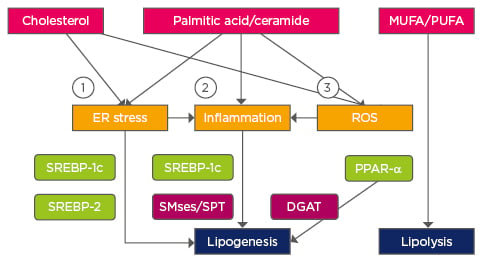
Figure 2: Pathways affecting lipid homeostasis in non-alcoholic fatty liver disease.
1) Ca2+ perturbation, ER membrane alteration; 2) TLR4; 3) Mitochondrial dysfunction.
Transcription factors are highlighted in green and enzymes in purple.
TLR4: Toll-like receptor 4; DGAT: Diglyceride acyltransferase; MUFA: monounsaturated fatty acids; PUFA: polyunsaturated fatty acids; ER: endoplasmic reticulum; ROS: reactive oxygen species; PPAR-α: peroxisome proliferator activated receptor alpha; SPT: serine palmitoyltransferase; SREBP: sterol regulatory element-binding protein; SMse: sphingomyelinase.
Further work needs to be done to fully elucidate NAFLD complexity,66 but it is clear that its inherent lipid accumulation translates into a lipid homeostasis dysregulation, with many lipid types being affected simultaneously and more importantly, many of them having specific biological functions.