Abstract
Chronic hepatitis B (CHB) is a serious consequence of hepatitis B virus (HBV), which infects and replicates in the liver. It is characterised by prolonged hepatitis B surface antigen seropositivity; this can lead to both cirrhosis and hepatocellular carcinoma (HCC). The infection begins when HBV binds its only known functional receptor, sodium taurocholate cotransporting polypeptide (NTCP), which was identified recently. The discovery of NTCP was a significant breakthrough in the field of HBV research, and has facilitated the establishment of a susceptible hepatoma cell line model for studying the mechanisms underlying HBV pathogenesis. Following productive HBV infection, both cellular and humoral immune cells and molecules, such as T cells and chemokines, are activated to resolve infection by destroying HBV-infected hepatocytes. However, host immunity to HBV is not always protective, most likely due to immune evasion mechanisms employed by HBV. These mechanisms may result in viral persistence, accumulation of mutations, and aberrant epigenetic alterations that lead to HCC. Here we highlight our current understanding of the HBV replication cycle, immunopathogenesis, and related mechanisms underlying the progression of CHB to advanced liver disease, along with the attendant complications.
INTRODUCTION
Chronic hepatitis B (CHB) results from hepatitis B virus (HBV), which belongs to the Hepadnaviridae family and the genus Orthohepadnavirus. CHB remains a significant contributor to morbidity and mortality related to sequelae such as cirrhosis and hepatocellular carcinoma (HCC).1 Recent statistics show that 240–400 million people have CHB worldwide, with the Pacific Islands, Africa, and Asia representing hyperendemic areas.2,3 More than 750,000 HBV-infected people die annually due to liver failure from cirrhosis and HCC. This still occurs despite the availability of HBV vaccines; recently, the FDA approved therapeutic agents such as nucleos(t)ide analogues and pegylated interferon, for use in the management of HBV.4
HBV is a prototype virus of 42 nm in diameter, with infectious double-shelled Dane particles that are indicated by the presence of hepatitis B surface antigen (HBsAg) in the blood. It is significantly pathogenic, leading to both acute and chronic infection.5 HBV is non-cytopathic, and therefore hepatic damage due to HBV infection results as a consequence of prolonged and aggressive activation of host immunity, aimed at eradicating the virus.5,6 Antiviral and antitumour immune responses are complex, with both the innate and adaptive systems affected and their dysfunction implicated in the progression of disease. More research efforts in understanding the early stages of HBV infection, host immunity, pathogenesis, and mechanisms underlying HBV progression to cirrhosis and HCC are urgently needed. This may lead to the development of therapeutic interventions that will elicit robust HBV-specific immune responses that clear the virus without damaging the hepatocytes. To this end, this review discusses research that elucidates mechanisms underlying HBV entry into the cell, immunopathogenesis, and hepatocarcinogenesis.
DISCOVERY OF THE HEPATITIS B VIRUS ENTRY RECEPTOR AND HEPATITIS B VIRUS REPLICATION
Horizontal, perinatal, and sexual transmission through contact with infected individuals and various bodily fluids are the predominant modes of HBV infection.7 HBV replication has been extensively studied over the last two decades; this led to the seminal discovery of the sodium taurocholate cotransporting polypeptide (NTCP) receptor, which facilitates HBV attachments and entry (Figure 1). NTCP, also known as SLC10A1, is abundantly expressed in the liver. It is involved in the transportation and clearance of bile acids from portal blood into hepatocytes. Yan et al.8 demonstrated that the preS1 region of the large protein L selectively interacts with the NTCP receptor following the binding of heparan sulfate proteoglycans, and this enables HBV attachment and entry into hepatocytes. HBV requires DNA polymerase and reverse transcriptase to replicate through RNA intermediates known as pregenomic RNA.5 Following entry into the hepatocytes, a HBV nucleocapsid is released into the cytoplasm and transported into the host cell’s nucleus to deliver relaxed circular (rc)DNA. In the nucleus, the rcDNA is repaired and converted to covalently closed circular super-coiled DNA, which serves as a template for the transcription of four viral RNA transcripts. The 3.5 kb pregenomic RNA transcript is then reverse-transcribed within the core particles. The resulting viral DNA within the core particles may either be enveloped and exported or recycled back to the nucleus, thus promoting HBV persistence in liver.6-11
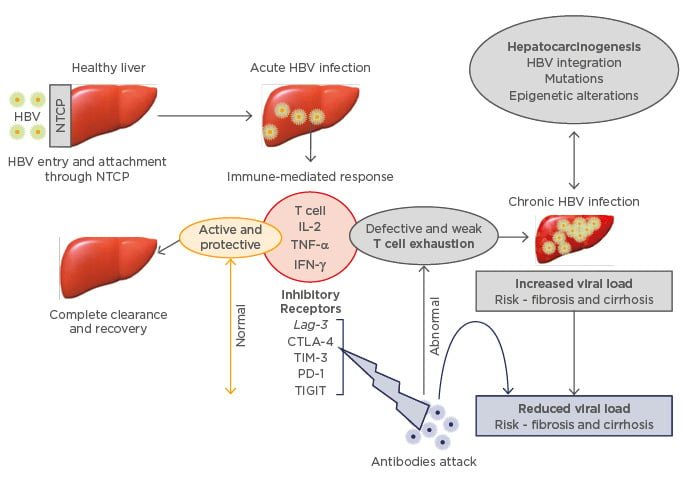
Figure 1: Impairment of T cell responses and cytokine disruption by the inhibitory cell receptors.
The effect on inhibitory receptors is associated with increased viral replication and aggressive liver disease. Blockade of the inhibitory receptors by specific antibodies restores T cell differentiation and cytokine production leading to resolved HBV infection with minimal liver damage.
HBV: hepatitis B virus; NTCP: sodium taurocholate cotransporting polypeptide; IL-2: interleukin 2; TNF-α: tumour necrosis factor alpha; IFN-γ: interferon gamma; Lag-3: lymphocyte activation gene 3; CTLA-4: cytotoxic T lymphocyte antigen 4; TIM-3: T cell immunoglobulin domain and mucin domain 3; PD-1: programmed cell death 1; TIGIT: T cell immunoreceptor with Ig and ITIM domains.
Knockdown of NTCP expression in duck primary hepatocytes infected with duck hepatitis B virus significantly decreased HBV infection, confirming that NTCP is indeed required for viral entry.12 Since the discovery of HBV, researchers have struggled to find an easily accessible model for studying HBV infection. It is now possible to achieve >50% infection efficiency in human-derived HepG2 and Huh7 cell lines expressing exogenous NTCP systems.12,13 Yan et al.14 showed that the spinoculation involving centrifugal inoculation of HepG2 cells expressing NTCP significantly enhanced HBV infection, which was dramatically reduced after treatment with HBV entry inhibitors such as irbesartan, which target NTCP.15 Even though further studies are urgently required to establish a model that will achieve 100% HBV-infection efficiency in in vitro or in vivo systems, the illustrated data support the evidence that NTCP is required for HBV attachment and entry. These findings provide new opportunities in HBV research for comprehending the early steps of HBV infection.
CHRONIC HEPATITIS B IMMUNE PATHOGENESIS
HBV is non-cytopathic, highlighting the complex but important interaction between the virus and host in causing HBV-related liver disease. Several studies on HBV transgenic mouse models have led to insightful knowledge on viral immunopathogenesis and the various strategies HBV utilises to escape immune responses.6,10 The liver is an immune-privileged organ as it is the site for both humoral and cell-mediated immunity involving resident macrophages (including Kupffer and natural killer cells) and CD4/CD8 T cells, which play significant roles in fighting against invading pathogens and malignant transformation.6
Acute HBV infection usually resolves spontaneously with robust production and activation of protective immune responses from neutralising antibodies, Kupffer cells, natural killer cells, CD4/CD8 T cells, interleukin (IL)-2, interferon gamma (IFN-γ), and tumour necrosis factor alpha (TNF-α) cells.16 Some individuals fail to clear the virus and progress to CHB infection; this is likely due to various factors including a long incubation phase before an immune response develops, an absence of ‘danger signals’ and viral evasion, resistance, deletion, alteration, and expansion of the immune system.17 Immune system anomalies are associated with immature dendritic cells and neutrophils, impaired production of cytokines (IFN-γ, IL-2, and TNF-α), and T cell exhaustion (the progressive loss of CD4/CD8 T cell function; Figure 1).17,18 These anomalies are attributable to HBV-mediated abnormal regulation of lymphocyte activation gene 3 (Lag-3), cytotoxic T lymphocyte antigen 4 (CTLA-4), T cell immunoglobulin domain and mucin domain 3 (TIM-3), programmed cell death 1 (PD-1) receptor, CD244/2B4, CD160, and T cell immunoreceptor with Ig and ITIM domains (TIGIT),19-21 which are outlined in Figure 1.
Immunoregulatory and inhibitory cell surface receptors are expressed on B cells, T cells, and cancer cells.19-21 PD-1 and CTLA-4 are the well described immunoregulatory receptors in CHB infection and cancer. The primary role of PD-1 is to minimise T cell activity in peripheral tissues at the time of an inflammatory response to infection. Upregulation of PD-1 was observed in CHB patients with elevated HBV DNA and alanine aminotransferase (ALT) levels, and this correlated with increased viral replication and active liver disease. Genome-wide microarray analysis in both acute and chronically HBV-infected mice also sheds some light by showing direct correlation of T cell impairment and upregulation of PD-1.22 Mechanisms underlying PD-1 activation in CHB infection have not yet been defined completely. However, it is known that PD-1 depends on its interaction with PD-L1 and PD-L2 ligands to negatively regulate T cell responses, cytokine induction, and proliferation.23,24 Most recently, it was shown that induction of PD-1 is partially regulated by endogenous transforming growth factor, which regulates T cell proliferation and differentiation.25
In cancer cells, upregulation of PD-1 promotes immune resistance by blocking activation of T cell receptor signalling pathways responsible for presenting invading pathogens to the major histocompatibility complex.18,25 One of the milestones that has recently emerged in the field of immunotherapy is the possible blockade of immunoregulatory receptors with specific antibodies to restore and enhance CD4/CD8 T cell immunity. Blockade of the inhibitory effect of PD-1 elicited robust production of HBV-specific CD8 T cell responses and resulted in intermediate T cell differentiation that significantly lowered viral replication, HBV DNA, and ALT levels in CHB patients.26,27 In cancer patients, PD-1 blockade resulted in tumour regression following inhibition of adaptive immune resistance.23 In a mouse model with productive HBV infection, the PD-1/PDL1 complex led to upregulation of intrahepatic PD-1 expressing CD8/CD4 T cell immunity, suggesting a strong correlation between upregulation of PD-1 and increased HBV.28 The use of anti-PD-1 antibodies in this mouse model blocked the interaction of PD-1 with PD-L1, leading to resolved HBV infection and reversal of the immune dysfunction. Li et al.29 and other studies corroborated these findings by showing that a combination of therapeutic vaccination and PD-1 blockade in chronic HBV infection enhanced T cell immunity, leading to resolution of CHB.29,30 Overall, these data suggest a new approach in the immunotherapeutic field to resolve CHB using PD-1 blockade and monoclonal antibodies.26,30
CTLA-4 is another significant immunoregulatory agent that becomes dysregulated during CHB infection. Several studies show that CTLA-4 negatively regulates T cell responses. In the early activation of HBV-specific CD8 T cells, induced by IFN-γ, CTLA-4 becomes upregulated, leading to increased levels of Bcl-2-interacting mediator of cell death, a novel member of the Bcl-2 family that promotes apoptosis via transcription of E2F-dependent mechanisms.19 A synergistic relationship between CTLA-4 with T cell receptor and CD28 impairs T cell responses by suppressing the production of IL-2 and T cell proliferation through activation of the apoptosis pathway.24,27 Blocking the CTLA-4 pathway results in the suppression of the apoptosis, leading to an increased expansion of IFN-γ-producing HBV-specific CD8 T cells.20 These may provide novel immunotherapeutic targets to eradicate CHB infection while minimising the risk of HCC progression.
CHB infection is characterised by persistent HBsAg seropositivity in infected individuals for >6 months.5 Laboratory and histological assessments through screening for the presence of HBsAg, hepatitis B envelope antigen (HBeAg), hepatitis B core antibody (anti-HBc), hepatitis B surface antibody (anti-HBs), HBV DNA, and ALT levels aid in differentiating the five clinical phases of CHB infection as illustrated in Figure 2. The division of CHB phases by the HBeAg status is clinically relevant, but is also indicative of the role of the HBeAg in inducing or regulating an immunological response.6 The first phase is immune tolerance, which is characterised by HBeAg seropositivity and normal ALT and liver histopathology, thought to be due to the tolerogenic effect of HBeAg. This was shown in the transgenic mice with deletion of specific T cell subsets and predominance of a Th2 response with the production of anti-inflammatory cytokines IL-4, IL-5, and IL-10.31
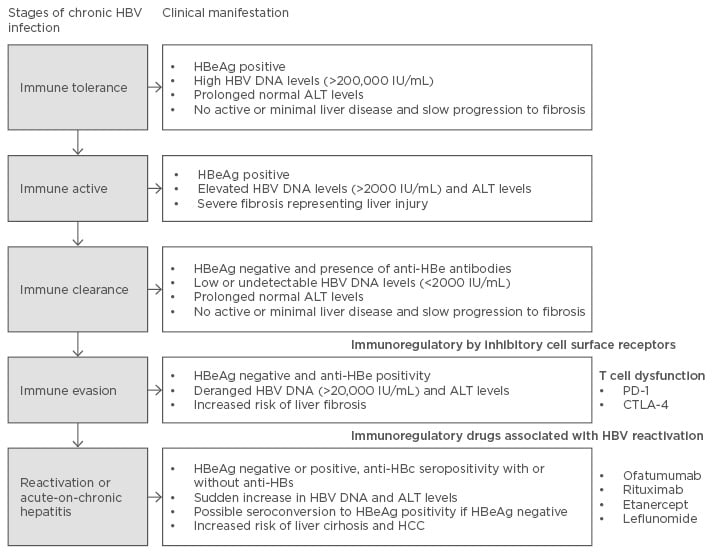
Figure 2: Chronic HBV replication phases and clinical manifestation.
HBV: hepatitis B virus; HBeAg: hepatitis B envelope antigen; ALT: alanine aminotransferase; anti-HBe: anti-hepatitis B e-antigen; HCC: hepatocellular carcinoma; anti-HBc: hepatitis B core antibody; PD-1: programmed cell death 1; CTLA-4: cytotoxic T lymphocyte antigen-4.
The second immune active phase is characterised by high HBV DNA and ALT levels, corresponding to the necroinflammatory activity seen in liver tissue associated with progressive disease. Immunologically, this phase corresponds to and is mediated by the secretion of proinflammatory cytokines, such as TNF-α, IFN-γ, and IL-20, and the increase in hepatitis C antigen-specific CD8 T cells seen in the liver. Contrastingly, seroconversion to HBeAg negative status and the appearance of hepatitis B envelope antibody (anti-HBe) in the serum is associated with decreased inflammation, suggesting the loss of the tolerogenic effect of HBeAg.31 Patients in the third phase of CHB, immune clearance, have anti-HBe antibodies and undetectable HBV DNA, normal ALT, and histology, associated with a lower but not eliminated risk of complications. Immune tolerance may be due to hyporesponsiveness of endogenous IL-12, and subsequent failure to induce HBV-specific memory CD8 T cell responses. The fourth phase involves immune evasion and is associated with increased risk of fibrosis, resulting from various factors such as viral integration (which disguise the virus from the immune system) and loss of antigenicity.6,31 The immune reactivation phase may occur with or without HBeAg expressing genomes, and is characterised by elevated HBV DNA and fluctuating ALT levels; these are associated with an increased risk of severe liver injury, which is likely explained by the prevailing proinflammatory cytokine milieu and reduction of CD8 T cells.31,32
Most recently, the FDA reported an increased fatal risk of HBV immune reactivation in patients receiving immunosuppressive therapy such as anti-CD20 agents ofatumumab and rituximab, which block the humoral immune responses by disrupting the function of B cells. Other immunoregulatory agents that may lead to HBV reactivation include etanercept, leflunomide, cyclosporine, and TNF-α inhibitors. It is therefore highly recommended that patients be tested for the presence of HBsAg, anti-HBc antibodies, and HBV DNA levels before they are treated with these immunoregulatory agents. Appropriate treatment of HBV must be immediately initiated in patients who develop HBV reactivation following treatment with ofatumumab and rituximab; ofatumumab and rituximab must be stopped immediately to prevent acute flares that may lead to liver transplant and death.33
UNDERLYING MECHANISMS OF HEPATOCARCINOGENESIS
Liver cirrhosis and HCC remain the major consequences of CHB. Globally, HCC is the third leading cause of global cancer-related deaths after colorectal and lung cancers.34 The available HBV therapeutic agents significantly reduce viral replication, but they still do not correct aberrant genetic and epigenetic alterations that promote progression of HBV to liver cirrhosis and malignant transformation of hepatocytes.35 The progression of HBV infection to HCC occurs in a series of steps generally following a sequence of CHB infection, fibrosis or cirrhosis, dysplastic nodule formation, and HCC development as illustrated in Figure 3. The average period of HBV infection progression to cancer is about 20–30 years, and 8–10 years after the development of cirrhosis.36-38
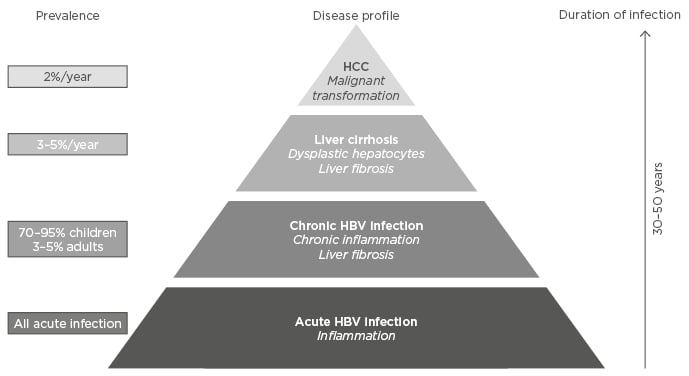
Figure 3: Chronic HBV infection, cirrhosis, and HCC development.
About 70–95% of children and 3–5% of adults infected with hepatitis B virus (HBV) may progress to chronic HBV infection associated with chronic inflammation and fibrosis.
HCC: hepatocellular carcinoma.
It is believed that over repetitive cycles of CHB infection, HBV DNA integrates into the host genome at preferential sites known as chromosomal fragile sites (e.g. FRA1A, FRA2C, FRA4E, etc.), which are frequent sites for translocation of chromosomes, oncogene amplification, and deletion of tumour suppressor genes in cancer.39 This increases the propensity for the accumulation of mutations and epigenetic alterations that may lead to fibrosis and ultimately cirrhosis and HCC development.39 Aberrant DNA methylation of promoter CpG islands is the primary epigenetic change seen during the course of HBV infection as it progresses to cirrhosis and HCC. Such methylation is detected at higher rates in HCC cells and less so in hepatocytes at the stage of CHB infection, fibrosis, and cirrhosis.40 In a study by Tseng et al.,38 high HBsAg levels were found to be associated with a risk of developing HCC even in the presence of low HBV DNA levels. This finding may be due to a higher degree of viral HBsAg integration into the host genome that would result in mutations and epigenetic alterations, particularly DNA methylation that affects both HBV and host genome.40
Evidence linking the DNA methylation process and CHB infection is extremely compelling. DNA methylation involves the attachment of a methyl group to the DNA coding sequence. This process is driven by the action of DNA methyltransferases, and may be classified as hypomethylation and hypermethylation.41 In CHB infection, integrated HBV DNA becomes methylated as part of the innate immune defence mechanism to alter viral gene expression, leading to repressed viral replication.40 For instance, toll-like receptors and IL-4, which form an important part of the innate immune system, were reported to be epigenetically regulated, leading to the activation of cellular pathways, such as the nuclear factor kappa B pathway, that result in reduced viral replication.42-44 Over time, the same methylation system that initially benefitted the host may start methylating the surrounding host promoter regions, causing aberrant activation or silencing in the transcription of genes including immunoregulators, tumour suppressors, and oncogenes, which are critical for the development of liver cancer.40
A large number of genes with distinct physiological functions have been found to be hypermethylated in the genome of patients with CHB or HBV-related HCC. These include classical genes such as RAR-β2, DLEC, IGJBP-3, LINE-1, RB1, ASPP1, E-cadherin, GSTP1, hTERT, caveolin-1, and p16INK4a genes. Aberrant methylation of these genes leads to silencing of gene transcription and perturbed cellular signalling pathways such as ubiquitination, DNA repair, proliferation, and apoptosis, which may lead to the development of HBV-related HCC.45-49
Although hypermethylation has been labelled as the key epigenetic regulator of gene transcription, other processes such as histone modification and microRNA (miR) expression have emerged as equally important in driving carcinogenesis. Genome-wide studies identified DNA methylation, histone modifications, and miR expression profiling across all the samples containing CHB and HBV-related HCC.50-52 However, amongst genome-wide studies there are discrepancies and data variations due to lack of proper, normal controls, heterogeneity of disease, variations in sample sources, and use of different technologies for analysis, suggesting the need for further validation.
Zhao et al.50 demonstrated that epigenetic alterations are multi-step events with unique profiling in different stages of progression from CHB to HCC development. MiR are small single-stranded RNA molecules that interact with messenger RNA targets to negatively regulate gene transcription.53 In HBV-related HCC, aberrant regulation of miR expression was observed and emerged as a potential indicator for diagnosis, prognosis, and therapeutic targets. For instance, in cirrhotic liver tissues containing underlying HBV-related HCC, miR-17-92, miR-17-5p, miR-21, miR-29, miR-34a, miR-93, miR-96, miR-100, miR-101, miR-122, miR-124, miR-199a/b, miR-222, miR-224, miR-425, and miR-529 have been aberrantly regulated.54-63 Distinct expression profiling of miRs is often associated with tumour development, aggressiveness, recurrence, metastases, and poor prognosis.61-63
Accumulated evidence indicates the significant role of epigenetic alterations in driving HBV pathogenesis and related carcinogenesis. Many studies have shown that epigenetic alterations are reversible42 and thus serve as potential targets for the development of markers for early diagnosis, prognosis, and therapeutic interventions.
FUTURE DIRECTIONS
Ample evidence highlights that CHB persists as a result of an impaired innate and adaptive immune system including immunoregulatory inhibitory cell receptors such as PD-1 and CTLA-4, which play a significant role in driving HBV progression to fibrosis, cirrhosis, and HCC. Several studies showed that the blockade of immunoregulatory inhibitory effects of cell receptors using antibodies restores T cell proliferation and cytokine production, leading to reduced viral replication and minimal inflammatory activity in the liver. Notably, epigenetic alterations could be considered part of the innate immune system aimed at suppressing viral replication. Unfortunately, similar epigenetic processes may also be hijacked and implicated in HBV hostile activities, resulting in the alterations of important genes, including immunoregulatory and tumour suppressors, which play critical roles in hepatocarcinogenesis. Future studies are required to enhance our understanding of the epigenetic consequences of HBV-host interactions in immunopathogenesis. This will aid in identifying novel potential immunotherapeutic targets that will help in the eradication of HBV infection and ultimately, progression to cirrhosis and hepatocarcinogenesis.