
Abstract
In the past decade, imaging has advanced to become a crucial tool in fundamental and biomedical research and it has become increasingly important to be able to image whole organs with single cell resolution. Light sheet fluorescence microscopy, also called selective plane illumination microscopy or ultramicroscopy, provides a high resolution in transparent and intact whole organs. By the application of a thin light sheet, only a defined slice of the specimen is illuminated and the fluorescence signal is detected by an objective perpendicular to the specimen. By moving the specimen vertically through the laser, a z-stack is acquired which corresponds to an optical sectioning without physical disruption of the specimen. The data can further be reconstructed to a three-dimensional volume and analysed in its entire complexity in micrometre resolution.
This article reviews the prerequisites for successful light sheet fluorescence microscopy, in terms of tissue preparation and optical clearing, and highlights recent advances and applications in the context of basic and biomedical research, with special focus on the central nervous system of rodents.
INTRODUCTION TO LIGHT SHEET FLUORESCENCE MICROSCOPY AND ESTABLISHED IN VIVO TOOLS
In biomedical research, as well as during the preclinical development of novel drugs and treatment regimens, in vivo and ex vivo imaging techniques have advanced to become major research tools used to address many important questions. A high spatial resolution is needed to investigate morphological changes and interactions at the cellular level, especially in preclinical disease models of the central nervous system (CNS), such as brain tumours, Alzheimer’s disease, or multiple sclerosis. Classical non-invasive in vivo imaging tools such as computed tomography (CT), magnetic resonance imaging (MRI),1,2 positron emission tomography, or single-photon computed tomography (SPECT),3 provide a spatial resolution in the sub-millimetre to millimetre range in living specimens. Yet, this resolution is not high enough to study single cells involved in disease pathology. In vitro and post mortem ex vivo microscopy techniques such as confocal or two-photon microscopy, which can also be applied in vivo, achieve a resolution of a few micrometres but involve the deterioration of the investigated sample required to be cut into thin slices prior to the imaging procedures.4 In the case of in vivo two-photon microscopy, a method that involves laborious sample preparations, clear subcellular resolutions of living organisms can be achieved.
However, the penetration depth is limited to ~200 µm (in vivo) to 500 µm (ex vivo), restricting the volume of investigation to the surface of the organ.5,6 Furthermore, problems of confocal and two-photon microscopy for in vitro and in vivo imaging are photobleaching and phototoxicity; optical clearing of organs allows for deeper penetration depths and when investigating cleared organs using light sheet fluorescence microscopy (LSFM), photobleaching and phototoxicity are of no concern.
In the past decade, LSFM has evolved to become the new imaging method of choice in biomedical research as it overcomes the issues of poor spatial resolution and limited penetration depth combined with the retained integrity of the specimen.7 In LSFM, the sample is illuminated by a thin light sheet perpendicular to the direction of observation. Moving the sample through the light sheet results in a stack of two-dimensional images; this can be referred to as optical sectioning. Importantly, the laser only illuminates the tiny sheet of a given specimen presently in focus. Therefore, the surrounding areas cannot outshine the imaged area and out of focus regions do not cause stray light.8 For the illumination of larger organs, such as whole mouse brains, two opposite lasers are commonly used for imaging. By using two light sheets, the sample can be uniformly illuminated without loss of light that would cause insufficient illumination and loss of information. Observation of samples often takes place in a liquid or gas environment. In ultramicroscopy (UM), whole organs can be used, ranging from several millimetres to centimetres in size. For a three-dimensional (3D) reconstruction of an organ or tissue using UM, hundreds or thousands of single images are collected in one measurement and create a picture of the whole specimen.9
The fundamental technique of LSFM was originally developed under the designation of UM by Richard Adolf Zsigmondy and Henry Siedentopf a century ago; it was awarded the Nobel Prize in 1925.10 The constant development of LSFM from the 1960s up until today’s powerful tool was comprehensively reviewed by Peter A. Santi.11 However, two major contributions were made by the groups of Ernst Stelzer,7 who invented the selective plane illumination microscopy (SPIM),12 and Dodt et al.,13 who applied optical clearing of specimens, paving the way for present applications. Today, this advanced technique represents a powerful tool to image total organs, whole animals (e.g. mouse embryos) or tissues and investigate the volume of interest in 3D reconstruction which allows for optical sectioning.14,15 The technique can also be used for in vivo applications in transparent specimens like fruit fly embryos or zebrafish. Other applications for LSFM are live imaging of 3D cell cultures, e.g. cellular spheroids, epithelial sub-organs, or stem cell organoids.14,16,17 Figure 1 illustrates a schematic description of a commonly used UM setup, showing the bidirectional laser excitation of the sample. Different filter settings can be applied for the excitation of the sample (Figure 1A). In this particular example derived from LaVision BioTec`s UltraMicroscope II, six filters can be incorporated into the microscope in order to measure different fluorescent signals in parallel. The objective enters the cuvette and detects the emitted fluorescence perpendicular to the sample (Figures 1B and C). With this setting, strong fluorescence signals from specimens of interest, in this case mouse brains (Figure 1C), are clearly detectable.

Figure 1: A schematic description of a commonly used ultramicroscopy-setup showing the bidirectional laser excitation of the sample.
Different filter settings can be used for the excitation of the sample (A). The objective enters the cuvette and detects the emitted fluorescence perpendicular to the sample (B and C). With this setting, strong fluorescence signals from the specimen, in this case from the mouse brain (C), are clearly detectable.
Copyright LaVision BioTec GmbH, Bielefeld, Germany.
Here, we will focus on the application of LSFM in biomedical research with optically cleared specimens. A recently published article by Pan et al.18 shows a newly developed clearing method allowing for imaging of entire adult mouse bodies.
OPTICAL SAMPLE CLEARING METHODS FOR LIGHT SHEET FLUORESCENCE MICROSCOPY APPLICATIONS
Optical clearing was initially invented by Werner Spalteholz in 1914, who adjusted the refractive index of the surrounding medium to the proteins of the specimen and obtained a transparent sample from that which was previously opaque.19 By adjusting the refractive index of the sample to the imaging solution, scattering of the laser is minimised and the light can cross the specimen with very little diffraction. During the last decade, several optical clearing methods were developed and improved. Becker et al.8,20-24 invented a multi-purpose clearing protocol for Drosophila, mouse embryos, mouse brains, and isolated mouse brain hippocampi. Today, various clearing methods with significant differences based on the solvents applied are used for tissue preparation, amongst others: ScaleA2,25 3DISCO,26 iDISCO,27 uDISCO,18 ClearT2,28 SeeDB,29 CLARITY,30,31 CUBIC,32 and FluoClearBABB.33 The common denominator of the clearing methods mentioned is to preserve endogenous fluorescence of proteins such as green or yellow fluorescent protein (GFP or YFP), red fluorescent protein from Discosoma sp. (DsRed) or mCherry expressed in the cells and organs of interest. In general, optical clearing of tissue by organic solvents is applied to match the refractive index of a tissue sample to a surrounding solvent. The first step of clearing involves the dehydration of the tissue since water has a lower refractive index than cellular structures like proteins and lipids.26 Afterwards the dehydrated tissue is impregnated with an optical clearing agent of the same refractive index. The tissue turns transparent and its composition is firmer than before.
Imaging of solvent cleared organs in 3D (3DISCO) and its successor techniques, whole-mount immunostaining and volume imaging (iDISCO) and ultimate DISCO (uDISCO), are frequently used clearing techniques to image neuronal connections in the nervous system.18,26,27 In order to get better clearing results of myelinated tissues in the adult CNS, 3DISCO was invented. Screening for a new chemical lead to the development of a new clearing protocol using dibenzyl ether, with the protocol for optical clearing of a mouse brain taking only 4–5 days.26
iDISCO is a simple, rapid, scalable, and inexpensive method for volume-imaging of whole-mount immunolabelled deep tissue structures. Existing whole-mount immunolabelling methods were tested and modified to achieve the deepest tissue penetration possible. Nearly 30 antibodies were shown to work well in immunohistochemistry and iDISCO.34 Glycine and heparin treatment was identified as a good option to reduce immunolabelling background in whole mouse embryos, whole adult mouse brains, kidneys, and other organs.27 The newly published uDISCO protocol is an improvement of 3DISCO to circumvent the disadvantage of quenching of endogenously expressed fluorescence signals. uDISCO uses diphenyl ether, an organic solvent with a refractive index of 1.579 in order to clear samples. In addition, Vitamin E is used to scavenge peroxides and tert-butanol, a dehydrating reagent that is more stable than the tetrahydrofuran used before in 3DISCO.18
The DISCO protocols enable high resolution imaging of neuronal connections within entire organs and even within an entire mouse without physical sectioning. The protocols for clearing are straightforward and can be performed in a relatively short time span of several days, depending on the size and tissue composition of the specimen. Subsequent imaging of the cleared specimen can be executed within several minutes to hours, depending on the scan region and scan protocol. However, a major drawback of the 3DISCO clearing protocol is the rapid loss of fluorescence. Hence, cleared samples need to be immediately scanned within 24 hours post-clearing. Due to the loss of fluorescence of 3DISCO-cleared samples, multiple scans of a region of particular interest can be challenging.26
The very recently published uDISCO method enabled imaging of whole adult rodents by taking advantage of the solvent-dependent shrinkage of tissue. The application of organic solvents for clearing causes a shrinkage of up to 65% of the original volume of the sample, which allows for imaging of undissected cleared adult mouse bodies. Importantly, the shrinkage does not affect microscopic or macroscopic scales.18
Another recently published clearing method, especially suited for imaging of whole adult mouse brains, is FluoClearBABB. Samples are initially dehydrated with an ascending series of butanols and the protocol requires 6 days for whole mouse brains. Afterwards, this method applies a mixture of benzyl alcohol and benzyl benzoate (BABB), in combination with an adjusted basic pH, which allows for whole brain clearing while reducing the optical distortion to a minimum.33 The majority of fluorescence is preserved for years with almost no photobleaching, enabling multiple repeated scans of the cleared specimens.
The CLARITY protocol, developed by the group led by Karl Deisseroth, is very different to the previously described clearing protocols.30,31,35,36 The method is based on an incubation of the tissue in a hydrogel matrix combined with an electrophoretical removal of lipids, a procedure that renders the specimen transparent. The CLARITY protocol was already used for the clearing of whole rodent brains and spinal cords,37,38 parts of the human brain,39 several other non-CNS organs from rodents, and also for clearing of embryos.40 This protocol preserves the structure of the cells, along with nucleic acids and proteins, and enables the localisation of RNA within the 3D specimen. However, the CLARITY protocol is very laborious and requires significantly longer time frames of weeks to months for the clearing procedure.
Another solution suitable for the clearing of whole brains, combined with preserved fluorescence, is the clear, unobstructed brain imaging cocktail (CUBIC) that achieves transparency of brains by the use of aminoalcohols.32 The CUBIC protocol is especially useful if the imaging of multiple fluorophores is desired.
LIGHT SHEET FLUORESCENCE MICROSCOPY APPLICATIONS IN BIOMEDICAL RESEARCH
Optical clearing can be applied to tissues or organs of any species, however, for imaging with LSFM the size of the specimen is restricted to several centimetres due to the design of the microscope. This limits its application mainly to mice and rats, if the investigation of whole organs is desired. A prerequisite for LSFM is the presence of fluorescence, either as protein or fluorophore, which labels the cell/region of interest. This can be done in various ways by the application of transgenic mice or xenografting of modified cells that express fluorescent proteins. Furthermore, the injection of fluorescently-labelled specific tracers, such as antibodies or proteins in general, ligands, or nanoparticles, is a frequently used method to visualise anatomical and subanatomical structures of interest, i.e. by the injection of fluorescently-labelled lectin that binds to endothelial cells to visualise the vasculature (Figure 2; Bode and Krüwel, unpublished data). Additional anatomical information can also be obtained by tissue-inherent autofluorescence. Figure 3 shows the utilisation of autofluorescence for LSFM imaging of a human cochlea. The sample was decalcified and cleared using FluoClearBABB. As already mentioned above, it is possible to stain dissected tissue post mortem with fluorescently-labelled antibodies to validate results.
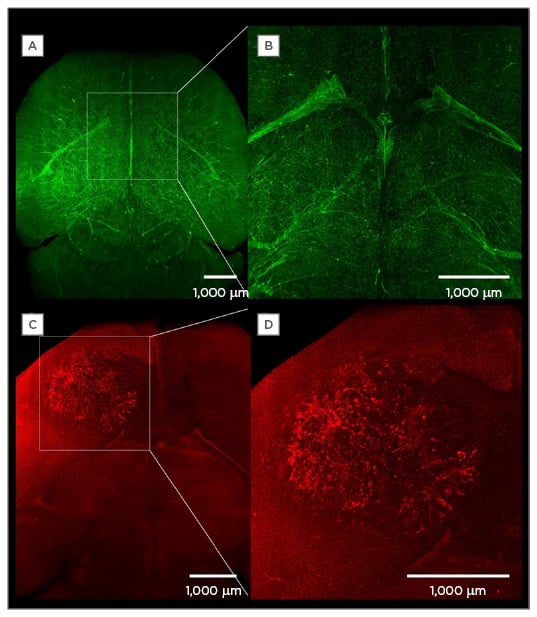
Figure 2: The injection of fluorescently-labelled tracers to visualise anatomical and subanatomical structures.
A) A maximum intensity projection of the vasculature of a healthy brain after intraveneous injection of fluorescently labelled lectin (12 mg/kg body weight) in the tail vein of a mouse; B) a magnification of A;
C) a maximum intensity projection of the vasculature of a mouse brain with a 3-week-old human U87 glioblastoma; D) a magnification of the tumour.
(Bode and Krüwel, unpublished data).
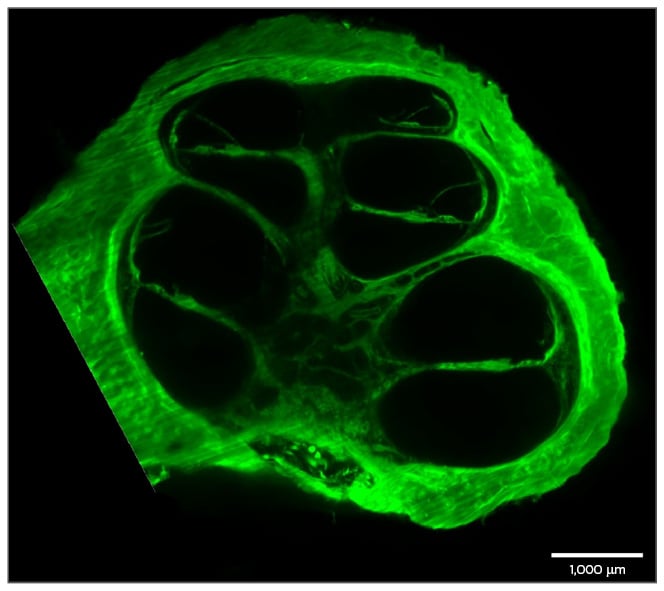
Figure 3: A preparation of a human cochlea.
The cochlea was decalcified and cleared using FluoClearBABB. Autofluorescence of the cochlea was then imaged using LSFM.
LSFM: light sheet fluorescence microscopy.
The sample was provided by Prof Dr M. Praetorius and M. Gestewitz, University Heidelberg, Heidelberg,
Germany; sample preparation and imaging were performed by J. Bode and T. Krüwel.
LSFM is a versatile technique for medical research. In basic research, the high resolution on a single cell level, in combination with the fact that the imaged specimens are intact and undissected, enabled novel insights into neuronal and vascular development patterns. Hägerling et al.41 applied LSFM on whole-mount immunostained mid-gestation mouse embryos to shed light on the development of the lymphatic system by precisely describing the morphogenetic events during the separation of the lymphatic from the venous endothelium. Belle et al.42 used LSFM to study axonal connectivity in transgenic mouse embryos and analysed axon guidance defects in the development of the neuronal system. Other studies have applied LSFM to investigate axonal regeneration and interaction of axons and scar-forming cells,43 or assessed the regeneration of optic nerves after injury, combined with the analysis of axonal trajectories.44 A very recent study described a workflow for the rapid acquisition of brain activity at cellular resolution by profiling immediate early gene expression, which highlighted the use of LSFM as a powerful platform for developmental biology.45 Additionally, LSFM was applied using a quantitative hydrogel-based technology to correlate activity in cells reporting on behavioural experience with measures for brain-wide wiring and regarding molecular phenotype.46 Stefaniuk et al.47 created a novel transgenic rat harbouring fluorescent reporter GFP expression under control of a neuronal gene promoter. This study is the first reference to a cleared rat brain, which exceeds the size of a mouse brain by far. The authors stated that FluoClearBABB clearing was found superior over passive CLARITY and CUBIC methods.
Besides these applications in developmental and behavioural biology, LSFM was extensively utilised for the investigation of diseases, especially those of the CNS, such as Alzheimer’s disease or brain tumours.48 In the case of Alzheimer’s disease, LSFM was used to assess the formation of amyloid plaques in whole mouse brains and in a part of the human brain.49 Surprisingly, the authors reported a higher complexity of the plaques of the human compared to the mouse brain. In translational biomedical research, LSFM helped to solve many different questions. Our group recently used LSFM to investigate the tropism and efficiency of adeno-associated viruses as transport vehicles for gene therapy of neuronal diseases.48 The use of undissected adult mouse brains allowed for a rapid 3D analysis of the viral transduction pattern of neuronal cells deep inside the brain. The high resolution of the microscope enabled the detection of single cells expressing the fluorescent proteins transduced by the viruses and proved the usability of this microscopic setup for higher throughput analysis. In glioblastoma, our group has investigated the role and modification of the vasculature during tumour progression and the effects of antiangiogenic treatments.50 Dobosz et al.9 and Weber et al.51 assessed the penetration of therapeutic antibodies in subcutaneous tumours and glioblastoma xenografts by LSFM.
THE EVOLUTION OF LIGHT SHEET FLUORESCENCE MICROSCOPY TO BECOME A POWERFUL TOOL IN BIOMEDICAL RESEARCH
A limitation of the method is the fact that only optically cleared tissue can be used for imaging and, thus, in non-transparent model systems, only ex vivo imaging is possible. By solely applying LSFM, dynamic monitoring of disease progression is laborious and involves a high number of animals. Therefore, our group and others combined LSFM with classical lower resolution in vivo imaging modalities, such as MRI and CT, to overcome this problem.50,52 By applying functional MRI (fMRI), the progression of glioblastoma models could be spatiotemporally monitored and the obtained data were registered to the high-resolution post mortem LSFM data.
An issue that needs to be solved in the future is the handling of the huge amount of data output obtained; one image is often >10 MB. Hence, full 3D image stacks easily exceed sizes of 10 GB. Therefore, better compression of these high-resolution images is desperately needed.
In this review, we presented LSFM as a novel and powerful imaging tool for basic and biomedical research and provided an overview of recent applications and requirements for imaging. If the interaction of single cells is the focus of research, LSFM is superior to all other microscopy techniques in terms of acquisition speed, imaging depth, sample size, photobleaching, and phototoxicity. The possibility to acquire a complete stack of images of an organ of interest without physical fragmentation will improve and accelerate biomedical research within the next few years.






