Abstract
Background: Human rhinovirus (HRV) is classified into three different species (HRV-A, HRV-B, and HRV-C), and several studies have reported a close relationship between the species and the severity of the disease. In a previous investigation, a high circulation of HRV was detected in paediatric patients from the city of Santa Fe, Argentina, hospitalised with acute respiratory infection.
Methods: A cross-sectional, retrospective study was conducted, consisting of the analysis of nasopharyngeal aspirate samples from hospitalised paediatric patients of Santa Fe city, from March 2010–February 2011. Clinical-epidemiological characteristics of the study subjects (N=56) were described. To characterise the circulating HRV species, RNA extracted from the samples was sequenced in both directions using specific semi-nested primers and automated analytical methods, after complementary DNA synthesis and reverse transcription PCR amplification. The phylogenetic tree was constructed by the maximum likelihood method using PhyML, with a topology search based on nearest neighbour interchange. The observed distribution was compared with studies that used the same molecular marker to characterise HRV sequences worldwide.
Results: Results showed HRV-A as the predominant species (66.1%), followed by HRV-C (21.4%) and HRV-B (10.7%). Notably, HRV-C infections were significantly associated with a history of asthma (p=0.024), asthmatic crises at discharge (p=0.024), and longer hospitalisation times (p=0.026). Phylogenetic analysis also identified four potentially novel HRV-C sequences forming a distinct clade.
Conclusion: This investigation successfully characterised the genetic diversity of HRV circulating in a paediatric population hospitalised with acute respiratory infection in Santa Fe. The findings highlight the clinical relevance of HRV-C in paediatric populations and its potential association with severe outcomes.
Key Points
1. Human rhinovirus (HRV) is a leading cause of acute respiratory infections in children, yet species-level surveillance data from Latin America remain scarce, limiting regional understanding of circulating viral diversity and its clinical implications.2. In a cross-sectional, retrospective study of 56 HRV-positive nasopharyngeal aspirates from paediatric inpatients in Santa Fe, Argentina (2010–2011), phylogenetic analysis was used to characterise circulating HRV species and assess associations with clinical outcomes.
3. HRV-A predominated (66.1%), but HRV-C was significantly associated with a prior history of asthma, asthmatic exacerbations at discharge, and longer hospitalisation times, supporting its role as a clinically relevant species in high-risk paediatric populations.
BACKGROUND
Human rhinoviruses (HRV), belonging to the genus Enterovirus and the Picornaviridae family, are spherical particles, non-enveloped and whose genome, a single linear molecule of positive sense RNA, is contained in an icosahedral symmetry capsid consisting of four viral proteins: VP1, VP2, VP3 and VP4. Thanks to the emergence of sequencing, which allowed the viral genome to be analysed in detail, variations in the structure of the capsid could be detected, particularly in VP1 and VP4/VP2, and this could also classify the HRV into one of the three already described species: HRV-A, HRV-B and HRV-C.1-3
Initially, it was thought that the only clinical manifestation associated with rhinovirus infection was the common cold, but this hypothesis was refuted with the development of molecular biology, which made it possible to detect the presence of this pathogen in infections of the lower respiratory tract4–7 and in patients with asthma.8-11 The relationship between the rhinovirus species and the severity of the disease has been reported in numerous research studies, where it has been concluded that the HRV-A and HRV-C species are more infective and related to severe pathologies.12-14
An annual study conducted in the city of Santa Fe, Argentina, revealed a high percentage of hospitalisations for acute respiratory infections (ARI) associated with rhinovirus and a percentage of positivity for this virus close to 40%, making it the second most circulated virus, behind respiratory syncytial virus (RSV).15 Due to the importance of these results, this work aims to characterise the diversity of circulating HRV species and genetically defined HRV types in that population, and to establish possible associations between rhinovirus species and the severity of clinical symptoms observed in previously studied paediatric patients.
METHODS
Type Of Study and Study Subjects
A cross-sectional, retrospective study was conducted, consisting of the analysis of nasopharyngeal aspirate samples from an original population of 99 patients under 15 years of age who were hospitalised in three hospitals in Santa Fe city, between March 2010–February 2011, with a diagnosis of ARI at the time of admission. All samples underwent RNA extraction, amplification, and sequencing procedures. Following sequence filtering and quality assessment, 56 RNA sequences were considered suitable for inclusion in the final analysis.
The studied samples were obtained from an original population of HRV-positive samples, provided by the National Institute of Respiratory Diseases “Dr. Emilio Coni” (INER), Santa Fe, and processed and diagnosed by Rudi et al.15 using a nested reverse transcription PCR (RT-PCR) that is highly sensitive and specific for most known genetically defined HRV types.
Sample Collection
Regarding the samples from the source population of the study by Rudi et al.,15 each participating healthcare facility was responsible for proper sample collection by the laboratory staff of the same institution, as well as for their storage and shipment for processing and analysis. The samples were labelled with a unique identification number (ID) for each patient and transported using a triple packaging system at a temperature of 4 °C to the laboratory of INER in the city of Santa Fe for processing.
Nucleic acids were extracted using silica membrane columns and the QIAamp Viral RNA kit (QIAGEN, Venlo, the Netherlands), and analysed for HRV using an RT-PCR assay developed by Steininger et al.,16 which amplifies a 93 bp fragment located at the end of the 5′ noncoding region of the HRV genome, conserved among all virus serotypes.
HRV-positive samples analysed in this study were obtained from an original population for which the absence of other respiratory viruses (RSV, adenovirus, and parainfluenza, as well as influenza A and B) had been previously confirmed by Rudi et al.15 In their work, Rudi et al.15 detected viral antigens for RSV, adenovirus, and parainfluenza using the indirect immunofluorescence technique, employing specific monoclonal antibodies and goat anti-mouse IgG conjugated with fluorescein isothiocyanate. Detection of influenza A and influenza B was carried out using real-time RT-PCR, following an in-house protocol transferred from the National Reference Laboratory, targeting the M gene and the NS gene, respectively. Samples with cycle threshold (Ct) values below 38 were considered positive.15
Although samples from the original population were collected from March 2010–February 2011, and HRV circulated throughout the entire period, the months of highest viral circulation were March–May 2010 (autumn), August (winter), and October–November 2010 (spring), with positivity rates ranging from 39.3–51.4% throughout the period.
Sample Inclusion Criteria
From the initial set of 99 HRV-positive samples, those sequences showing ≥90% identity with reference HRV sequences in the NCBI nr database with BLAST algorithm17 were retained. Finally, 56 HRV-positive samples were included in the final analyses. This criteria is consistent with the HRV molecular classification, where sequence identity values of approximately 87–90% are commonly used to support genotype assignment and phylogenetic clustering, particularly in the VP4/VP2 and VP1 regions.18,19
The remaining samples (n=43) were excluded due to identity lower than the predefined threshold, low sequence quality, and/or insufficient genomic coverage, which may indicate non-specific amplification or unreliable sequence data. Given the high genetic diversity of HRV and the extensive nucleotide variability observed among circulating strains, the application of stringent sequence identity thresholds is particularly important to ensure reliable taxonomic assignment and to minimise the inclusion of non-specific or low-quality sequences.20
Although this filtering reduced the sample size, it ensured accurate species classification and robust phylogenetic inference, minimising the risk of misclassification bias in a highly heterogeneous RNA virus.
Clinical Characteristics of the Subjects
Following the implementation of these sequence quality and taxonomic reliability criteria, the resulting cohort of 56 HRV-positive cases was further examined to describe its demographic and clinical characteristics: 53.6% were male and 46.4% were female; 60% were younger than 6 months and more than 80% were younger than 5 years. In 77.0% (43) of the cases, no clinical history of risk factors or previous diseases was reported, whereas five cases had a history of prematurity, three of congenital heart disease, two of asthma, and one of malnutrition. The most frequent discharge diagnosis was bronchiolitis (35.7%), followed by pneumonia (25.0%) and bronchitis (14.3%).
Amplification and Sequencing of Extracts
RNA extracted from samples where the rhinovirus genome was previously detected, was used in this investigation. The typing was performed by amplifying and sequencing a 542 bp fragment of the viral genome containing the VP4/VP2 regions, using a reverse primer and external and semi-nested forward primers obtained from previous research.21
The complementary DNA synthesis was performed by RT-PCR using the OneStep RT-PCR kit (QIAGEN). The final reaction volume was 25 µL and contained 5 µL of buffer (5 X), 1 µL of deoxynucleoside triphosphates (10 µM), 2.5 µL of external forward and reverse primers (5 µM), 1 µL of enzyme mix OneStep (QIAGEN), 5 µL of RNA, and water to complete volume. The incubation steps for the reverse transcription process were initially 30 min at 50 °C, followed by 15 min at 95 °C and finally, 2 min at 95 °C. Then, 35 PCR cycles were carried out under the following conditions: 30 sec at 95 °C, 30 sec at 45 °C, and 30 sec at 72 °C. As a final step, a final extension was carried out at 72 °C for 5 min. The amplification of the final product was carried out in a second PCR, whose final reaction volume was 50 µL and which contained 5 µL of buffer (10 X) with MgCl2 (20 mM), 2 µL of deoxynucleoside triphosphates mixture (10 mM), 1 µL of semi-nested forward and reverse primers (5 µM), 1 µL of DreamTaq FERMENTAS enzyme (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA; 5 U/µL), and 2 µL of the complementary DNA obtained in the previously performed PCR. The PCR product was visualised on an agarose gel (2%).
The concentrations of the amplicons were determined using the NanoDrop™ Lite spectrophotometer (Thermo Fisher Scientific inc.) and the corresponding dilutions were made to adjust them to an optimum final concentration for their sequencing (10–20 ng/µL). The sequencing was performed in both directions with the semi-nested forward and reverse primers using an ABI 3730XL Sequencer (Thermo Fisher Scientific Inc).
Phylogenetic Analysis
Trev22 was used to edit the 56 RNA sequences from their corresponding electropherograms, eliminating the ends that did not have a correct base calling (phred number <20). Then, a multiple sequence alignment was performed with MAFFT v7.40223 using the 56 sequences of interest. This alignment was edited with JalView24 deleting the misaligned ends. Finally, with the maximum likelihood method using PhyML,25 the phylogenetic tree was constructed, and using 1,000 replicate bootstraps as a branch technique (tree support or reliability), a topology search was chosen for the exchange of nearest neighbors (NNI: nearest neighbor interchange). The phylogenetic tree graph was generated with iTOL.26 The taxonomic assignment of the 56 RNA sequences was performed using BLAST and Genome Detective 27
HRV Distribution
This distribution was compared with studies that used the same molecular marker to characterize the Rhinovirus sequences in different locations of the world, taking into account that these studies are subsequent to the year 2008, when the HRV-C species was defined.
The coefficient of variation28 was used to determine if the distribution percentages of the rhinovirus species in this study are similar to those performed by others in different locations, as observed in Table 1.
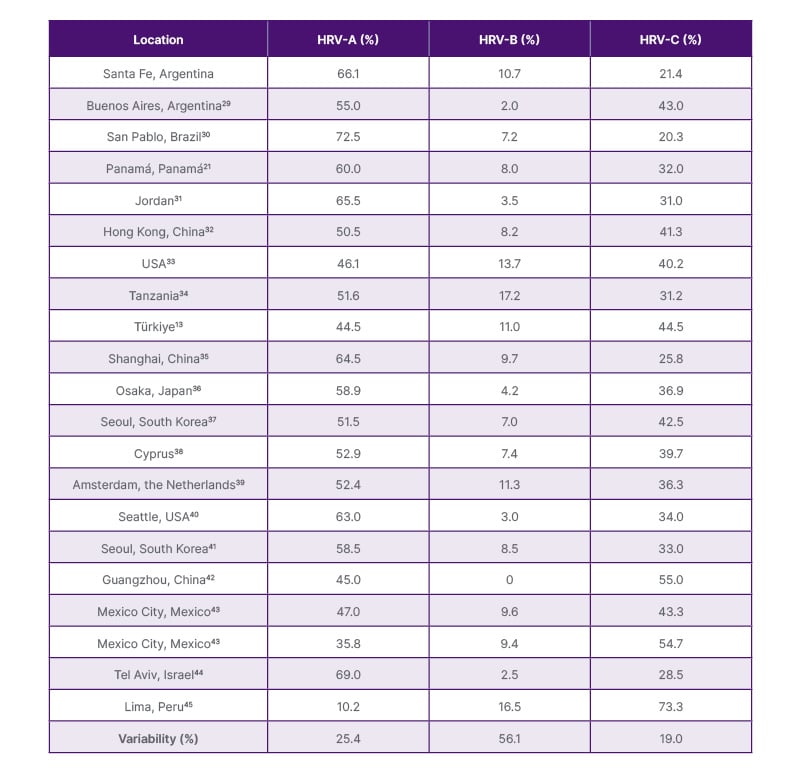
Table 1: Distribution of rhinovirus sequences in different populations around the world.
HRV: human rhinovirus.
GC Content
The GC content of each sequence was calculated for both the analysed sequences and the reference sequences with the geecee tool of the bioinformatic package EMBOSS. Then, the average GC content, with its respective standard deviation, was obtained for each rhinovirus species.
Statistical Analysis
Statistical analysis of clinical and epidemiological data was performed to explore potential associations between HRV species and patient characteristics and outcomes. Fisher’s exact test was applied for categorical variables given the small subgroup sizes, and the Mann–Whitney U test was used for continuous variables. The student’s t-test or non-parametric Mann-Whitney U test, ANOVA tests, or Kruskal–Wallis test were used for continuous variables, when applicable. Any p values <0.05 were considered as statistically significant.
Ethical Considerations
The present study was reviewed and approved by the Provincial Bioethics Committee of the Province of Santa Fe (Provincial Registry No. 471/2015) on 18th August 2015. In addition, authorisation was obtained from the executive boards of the participating hospitals and from INER prior to the start of the study.
Data handling was conducted in accordance with Argentine National Law No. 25,326 on the Protection of Personal Data. All data were consolidated into a database without personal identifiers, using numerical coding.
RESULTS
Assignment of Species and Types
The construction of a phylogenetic tree (Figure 1), was used to define the assignment of the sequences analysed at the species and type level together with the search carried out with BLAST and Genome Detective.
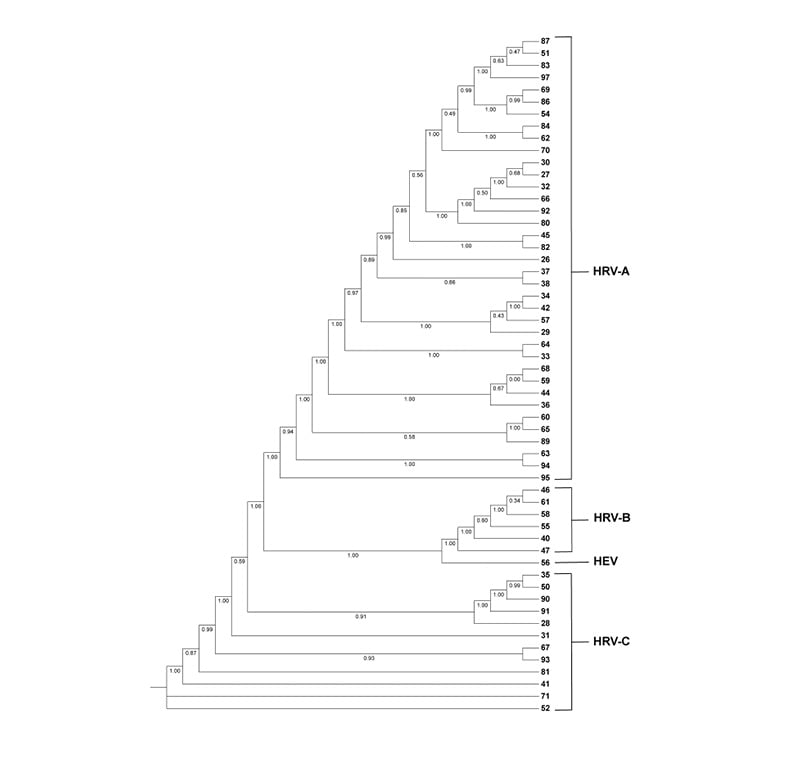
Figure 1: Phylogenetic tree of rhinovirus sequences distributed in the HRV-A, HRV-B, and HRV-C clades, along with a sample belonging to the HEV clade (n=56). The numbers on the leaves of the phylogenetic tree correspond to the ID of each RNA sequence.
HEV: human enterovirus; HRV: human rhinovirus.
All sequences were assigned at the species level: 37 sequences were assigned to the species HRV-A (66.1%), six to the species HRV-B (10.7%), 12 to the species HRV-C (21.4%), and one to the HEV species (1.8%). Moreover, at type level, six sequences (10.7%) were not able to be assigned to a particular HRV type, three of them corresponding to the species HRV-A, one corresponding to the species HRV-B, and two to HRV-C clade.
The HRV-A strains for each genotype were as follows: A8 (1); A10 (3); A12 (4); A15 (1); A21 (1); A22 (1); A24 (4); A29 (2); A30 (1); A40 (3); A46 (2); A49 (1); A51 (2); A57 (1); A65 (2); A78 (1); A81 (2); A82 (2). The present HRV-B strains in each genotype were as follows: HRV-B3 (1); HRV-B14 (1); HRV-B83 (2); HRV-B91. Finally, the HRV-C strains for each genotype were as follows; HRV-C6 (1); HRV-C7 (3); HRV-C12 (1); HRV-C26 (1); HRV-C39 (1); HRV-C41 (1); HRV-C42 (1); HRV-C43 (1). Notably, six sequences could not be assigned to a specific HRV type despite successful species-level classification. These included three HRV-A sequences (ID 83, 89, and 97), one HRV-B sequence (ID 58), and two HRV-C sequences (ID 52 and 71), suggesting potential genetic divergence from currently recognised reference types.
Distribution of Rhinovirus Sequences in Different Locations
When comparing the estimated distribution percentage for each species (66.1% for HRV-A, 10.7% for HRV-B, and 21.4% for HRV-C) with findings from different locations worldwide, variations of 12.0%, 45.0%, and 19.0% were obtained for the HRV-A, HRV-B, and HRV-C species, respectively (Table 1).
GC Content
The GC content for the sequences of the species HRV-A was 40±1.6, for the species HRV-B was 41±1.1, and for the species HRV-C was 44±1.9.
Rhinovirus Species and Clinical Associations
Table 2summarises the clinical and epidemiological characteristics of patients with a positive diagnosis of HRV, stratified according to the detected HRV species. Overall, no significant differences were observed in mean age or sex distribution among the groups. However, statistically significant differences were identified in some clinical variables. HRV-C infections were significantly associated with a prior history of asthma (Fisher’s exact test, p=0.024) and with an asthmatic crisis recorded at discharge (Fisher’s exact test, p=0.024). Patients infected with HRV-C had a longer median hospitalisation time (12.4 days) compared to those infected with HRV-A or HRV-B (Kruskal–Wallis test, p=0.026). No significant differences were observed in age, sex distribution, oxygen requirement, fever, ICU admission, or discharge diagnoses across the three species groups (all p >0.05; Table 2). All statistical analyses were exploratory and unadjusted for multiple comparisons. These associations should therefore be interpreted as preliminary and hypothesis-generating, given the limited number of HRV-C cases (n=12) and the very small number of asthma-related observations (n=2), which preclude any causal inference or generalisable conclusions. In addition, both patients with a documented history of asthma in this cohort were infected with HRV-C. Although the number of cases is limited, this observation is consistent with the statistical associations described above and further supports a potential link between HRV-C infection and asthma-related clinical outcomes.
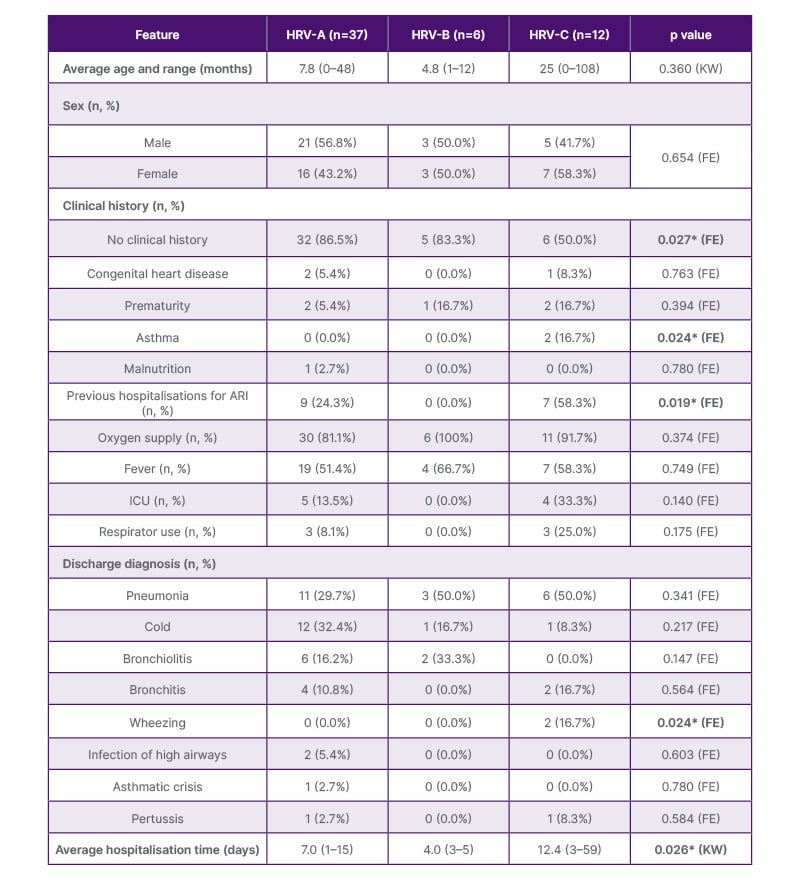
Table 2: Clinical-epidemiological features of patients with a positive diagnosis of rhinovirus and acute respiratory infections.
*Statistically significant (p<0.05).
All analyses are exploratory and unadjusted for multiple comparisons.
Note: Percentages for sex and discharge diagnoses are calculated over the total per species group. Percentages for clinical history sub-categories are calculated over the total of each species group (n). Low-frequency categories (n≤2 in all groups) should be interpreted with caution.
ARI: acute respiratory infection; FE: Fisher’s exact test; KW: Kruskall–Wallis test. (Continued)
DISCUSSION
Types Assignment
The presence of several sequences that could not be confidently assigned to currently recognised HRV types suggests a level of genetic divergence that may reflect the high evolutionary dynamics of rhinoviruses. This could be due to the exceptionally high nucleotide mutation rate that RNA viruses exhibit, which is also observable on a timescale comparable to the host immune system’s response capacity.46 This excellent adaptability and high genetic heterogeneity also lead to phenotypic variability in the population, where physical fitness is not uniformly distributed among all coexisting strains.47
The observed association between HRV-C infection and asthma-related variables is consistent with previous reports suggesting increased susceptibility of asthmatic airways to HRV-C and a potential role of this species in triggering more severe exacerbations.48,49
Notably, four of the unassigned HRV-C sequences (IDs 52, 71, and two additional sequences forming a distinct clade in the phylogenetic tree) appeared to cluster separately from all currently recognised HRV-C types, which could suggest the presence of potentially novel HRV-C types in circulation. However, this interpretation must be expressed with caution. Formal designation of a novel HRV type requires meeting specific criteria established by the International Committee on Taxonomy of Viruses (ICTV) and described by McIntyre et al.,19 including sufficient pairwise nucleotide distance from all recognised types in the VP1 region and robust phylogenetic support. The VP4/VP2 fragment analysed in this study provides a useful initial classification tool, but is insufficient on its own for definitive novel type assignment. Full-length VP1 sequencing or whole-genome characterisation would be needed to substantiate this claim. Accordingly, these four sequences are best described as ‘putative novel types’ or ‘unclassifiable sequences warranting further characterisation’, rather than formally proposed novel HRV-C types.
GC Content
The GC composition is an important genomic factor that can be evolutionarily optimised for adaptation to multiple environmental constraints (such as ideal growth temperature).50
GC content is an identifying mark within an organism. In addition, as an important feature of the mating of the GC bases, their greater thermal stability is highlighted by forming a triple hydrogen bridge, in comparison with the AT bases that form two hydrogen bonds. Even so, the thermal stability occurs not only because of the three hydrogen bonds that are formed between the GC bases, but also because of the quantity of these arranged consecutively.51
The observed GC content patterns indicate that HRV-C sequences tend to exhibit higher GC content compared to HRV-A and HRV-B, in agreement with previous reports.50,52 The GC content does not seem to be the most adequate strategy to differentiate Rhinovirus species since, despite having a clear distinction between HRV-A and HRV-C species versus HRV-C, it is very difficult to differentiate within this value at the species HRV-A and HRV-B for being very similar. On average, the sequences of the HRV-C species show a higher GC content than the sequences of the other two species.
Distribution of Rhinovirus Sequences in Different Locations
The relatively consistent distribution of HRV species across geographic regions suggests that global circulation patterns are largely conserved, with HRV-A typically predominating, followed by HRV-C and HRV-B. This observation aligns with previous large-scale analyses, supported by the review published by Esneau et al.,53 in 2022 where, when studying 31 articles, they observed that HRV-A species was the most detected overall (56% on average; minimum of 44% of detection and a maximum of 75.9% of detection). The next most detected species was HRV-C, with a minimum of 20% of subtypes belonging to this species and a maximum of 55% (the average was 34.5%). Although 19% of HRV subtypes belong to HRV-B species, the maximum of detection was 18.2% in one study and 8.5% on average for this species.54
Limitations
Regarding the limitations of this study, it should be noted that the cross-sectional and retrospective design precludes causal inference; therefore, the observed associations between HRV species and clinical outcomes should be considered exploratory and hypothesis-generating.
Second, the overall sample size was modest (n=56 after quality filtering), and subgroup sizes were particularly small, which limits the statistical power of subgroup comparisons and, therefore, these findings should be interpreted with caution.
Additionally, both the historical sampling period, as well as the duration (12 months) and the study setting (a single city), may not reflect current patterns of HRV circulation nor capture interannual and regional variation in viral diversity. In this regard, it would be of interest to replicate the study under current conditions, over a longer period and across different regions of the country.
Although HRV-positive samples were confirmed negative for RSV, adenovirus, parainfluenza, and influenza A and B, other potential viral or bacterial co-pathogens were not systematically evaluated. The possible presence of undetected co-infections represents a limitation of this study, as such agents could influence clinical severity; however, given the absence of systematic co-infection data, no conclusions regarding their confounding effect can be drawn.
Finally, it should be considered that this study predates the widespread adoption of more comprehensive sequencing approaches; although standard for HRV typing, there are limitations regarding the definitive assignment of novel types and full genomic characterisation.
CONCLUSION
This investigation successfully characterised the genetic diversity of HRV circulating in a paediatric population hospitalised with ARI in Santa Fe, Argentina. The analysis of 56 samples revealed that HRV-A was the predominant species (66.1%), followed by HRV-C (21.4%) and HRV-B (10.7%). This distribution aligns with global trends, showing that HRV-A is consistently the most prevalent species worldwide, irrespective of geographical location, while HRV-B remains the least common.
The statistical analysis of clinical data suggested potential associations between HRV species and patient outcomes. In particular, HRV-C infections were more frequently observed in patients with a history of asthma (Fisher’s exact test, p=0.024) and in those who experienced asthmatic exacerbations at discharge (Fisher’s exact test, p=0.024). Additionally, patients infected with HRV-C showed a longer average hospitalisation time (12.4 days) compared to those with HRV-A or HRV-B (Kruskal–Wallis test, p=0.026). All analyses were exploratory and unadjusted for multiple comparisons.
Due to the relatively small overall sample size, limited subgroup sizes, and the cross-sectional retrospective study design, along with the absence of adjustment for multiple comparisons, causal inferences cannot be established. In addition, the potential confounding effect of unmeasured co-infections cannot be ruled out, as only a limited set of respiratory viruses was assessed. Consequently, although these findings are consistent with previous literature suggesting greater clinical severity associated with HRV-C, particularly in vulnerable populations such as children with asthma, conclusions regarding its clinical impact should be interpreted with caution.
The observed GC content further distinguished HRV-C, which exhibited a significantly higher average GC content (44%) compared to HRV-A (40%) and HRV-B (41%), providing a genomic marker that corroborates the phylogenetic classification.
In conclusion, although it would be valuable to compare these findings with more up-to-date data on circulating strains, this study confirms the high circulation of HRV, particularly HRV-A and HRV-C, as significant pathogens in paediatric ARI in Santa Fe. The observed associations between HRV-C infection and asthma history, asthmatic exacerbations, and longer hospitalisation suggest a potential link with greater clinical severity.
Although these findings should be interpreted as exploratory, they support existing evidence and underscore the need for continued surveillance and further research to better characterise the clinical impact and pathogenesis of HRV-C, particularly in high-risk paediatric populations. Future studies incorporating larger sample sizes and prospective, multicentre designs, along with multivariable analyses, will be essential to validate and extend these findings and to more accurately assess the independent contribution of HRV species to clinical outcomes, while accounting for potential co-infections, ultimately informing clinical management strategies. In addition, studies incorporating viral load quantification and longitudinal designs will be essential to better define the relationship between HRV species, viral dynamics, and clinical outcomes. Finally, the application of more extensive sequencing approaches could deepen the understanding of genetic diversity and potential virulence determinants, particularly in HRV-C. In this regard, the identification of potentially novel sequences in this study highlights its contribution to the understanding of viral diversity and underscores the importance of continued genomic surveillance.




