
Meeting Summary
With new patient-centric and scientific networks being created, Prof Chorostowska-Wynimko explored how these initiatives, such as the European Alpha-1 Research Collaboration (EARCO) and the European Reference Network-LUNG Alpha1 Antitrypsin Deficiency (ERN-LUNG AATD Core Network), will help to advance the management of alpha 1-antitrypsin deficiency (AATD) patients. EARCO plans to create a registry to gather information from centres across Europe and ERN-LUNG AATD plans to ensure highly specialised healthcare for AATD patients, including reliable AATD diagnostics across European laboratories.
Explaining in more detail the plans for the new EARCO registry, Dr Barrecheguren argued the case for another AATD registry to gather large-scale data that clinical trials cannot provide. She provided an overview of the new EARCO prospective follow-up registry, to be launched next year, which will integrate existing national AATD registries, enhance long-term follow-up and quality of data, and facilitate research and quality improvements across healthcare systems.
Discussing one of the first initiatives of the ERN-LUNG AATD Core Network, Dr Ferrarotti explored how to align AATD testing across Europe with the creation of European LAB-NET, an initiative first involving six European centres that will co-operate to collect, develop, verify, and make reference materials available for molecular and biochemical tests to correctly diagnose AATD and provide quality control in the laboratory diagnosis.
Dr Greulich reported on a post-hoc pooled analysis from the RAPID-randomised controlled trial (RAPID-RCT) and the RAPID-open label extension (RAPID-OLE) study, which compared the safety and tolerability of adverse event (AE) rates for two different alpha-1 antitrypsin (AAT) dosing patterns, weekly infusions of 60 mg/kg AAT, and bi-weekly infusions of 120 mg/kg AAT. Results showed there were no significant differences for exposure-adjusted event rates (p=0.850), infusion-adjusted event rates (p=0.344), and serious treatment emergent AE (TEAE) (p=1.0); TEAE occurring in the first 24 and 48 hours were comparable for both groups.
Prof Sandhaus presented the results of a telephone survey from the USA AlphaNet organisation of self-infusion practices in 555 patients with AATD. The survey found that 7.9% of respondents self-administered AAT and 92.1% who did not. Of the 44 patients who self-administered AAT, 95.4% reported being very satisfied and 4.6% were satisfied with their treatment.
Rare Lung Diseases in The Spotlight: New European Initiatives
Professor Joanna Chorostowska-Wynimko
In 2012, a group of experts evaluated barriers to accessing therapies for rare lung diseases in Europe and called for greater collaboration and communication between stakeholders (patient groups, physicians, drug developers, regulators, and payers) and better integration of research across countries and continents to address existing challenges.1
As one example as to why better collaboration is dearly needed, Prof Chorostowska-Wynimko presented an example from epidemiological research. An epidemiological study of distribution of AATD published in 2004 reported the highest frequency of the PI*Z variant in northern and western European countries, while the highest frequency of the PI*S variant was described for southern European countries.2 For both variants, the lowest numbers occurred in Eastern Europe, with a sharp drop occurring along the former iron curtain.2 However, this cannot be considered to be a true reflection of the frequencies of deficiency alleles, but rather a result of the disparities in the medical care between Eastern and Western Europe. A 2017 updated analysis of PI*S and PI*Z frequencies no longer showed this East–West divide.3 Such disparities emphasise the need for better co-ordinated, good-quality research and care for AATD patients throughout Europe.
In April 2018, the European Respiratory Society (ERS) launched the European Alpha-1 Research Collaboration (EARCO) to build a network of AATD researchers and clinicians to guide clinical, research, and translational priorities in accordance with patients’ concerns. The initiative comes under the ERS Clinical Research Collaborations umbrella, which supports projects in different respiratory areas with the objective of building and maintaining European multicentre research networks. EARCO has the additional purpose of creating the European AATD EARCO registry to ensure long-term follow-up, facilitate patient recruitment for research, and improve quality initiatives across healthcare systems. EARCO is chaired by Dr Marc Miravitlles (Spain) and Dr Timm Greulich (Germany) and is run by a steering group of researchers and patient representatives.
The above-mentioned 2012 expert statement identified the need to use a limited number of patients in rare diseases wisely, learn from the model of the Rare Lung Diseases Consortium in the USA, and write consensus documents. With such objectives in mind, the ERS published a statement in 2017 on the diagnosis and treatment of AATD that outlined optimum care provisions in an integrated management programme.4 Management programme recommendations included primary diagnosis with AAT genotypes and phenotypes, confirmation of AATD diagnosis, disease staging, and properly structured patient follow-up. To help establish such a disease management programme is the major aim of the AATD Core Network that was recently created within the ERN-LUNG.
ERN-LUNG is a non-profit network of European healthcare providers committed to the prevention, diagnosis, and treatment of rare respiratory diseases through patient care and advocacy, education, and research. The AATD Core Network is one of the nine Core Networks within ERN-LUNG, with AATD core centres located in Leiden, Netherlands; Warsaw, Poland; and Pavia, Italy, and supporting partners in Dublin, Ireland; Barcelona, Spain; Homburg, Germany; Birmingham, UK; and Leuven, Belgium. Other ERN-LUNG Core Networks exist for cystic fibrosis, pulmonary hypertension, primary ciliary dyskinesia, non-cystic fibrosis bronchiectasis, mesothelioma, chronic lung allograft dysfunction, and other rare lung diseases.
Benefits of an AATD management programme in the USA have been reported in a number of publications.5-7 Data from the Alpha-1 Disease Management and Prevention Program (ADMAPP) in the USA comparing patients adhering to the programme (n=1,605) with those who did not (n=1,921) found that adhering patients were more likely to have had a pneumonia vaccine in the last 6 years (p=0.017), flu vaccine in the last year (p<0.001), hepatitis A vaccine ever (p<0.001), hepatitis B vaccine ever (p<0.001), and to exercise (p<0.001).5 In a second analysis comparing the outcomes for 878 AATD patients, both 12 months before enrolment in ADMAPP and 12 months after, patients showed improvements in mean number of exacerbations (p<0.001), mild exacerbations (p=0.006), moderate exacerbations (p<0.001), and never having experienced an exacerbation (p=0.003).6 The disease management programme was also cost-effective. Comparing 213 AATD patients assigned to the health management programme with 232 AATD patients not assigned found that the mean total costs were \$142,406 for the managed programme cohort versus \$167,935 for the comparator cohort (p=0.010), and that the medical costs were \$113,051 for the management programme cohort versus \$138,045 for the comparator cohort (p=0.021).7 So far, no disease management programme for AATD in Europe exits. The ERN-LUNG Network recently launched a patient management platform, the Clinical Patient Management System, to facilitate live clinical case discussions between members and enable cross-border consultations.
Future plans include establishing a quality validation programme for standardisation of AATD diagnostics, a network of certified laboratories, and close collaboration between the AATD Core Group and EARCO CRC (that is, the creation of an AATD registry). Initiatives providing good-quality laboratory data should be given the highest priority since they are fundamental for the accurate identification of AATD patients. The ERN-LUNG AATD Core Network also aims to closely collaborate with EARCO and the AATD registry and include strong patient involvement.
Do We Need Another Alpha-1 Antitrypsin Deficiency Registry?
Doctor Miriam Barrecheguren
With an estimated prevalence of 20 people per 100,000 of the population, AATD affects more patients than other well-known rare pulmonary diseases, such as pulmonary arterial hypertension, interstitial lung disease, cystic fibrosis, idiopathic pulmonary fibrosis, and sarcoidosis.8 AATD is significantly under-diagnosed,9 with an average delay in diagnosis of 5.1 years from first presentation of symptoms.10 A Spanish primary care study demonstrated that the number of AATD determinations was low in relation to chronic obstructive pulmonary disease (COPD) prevalence. Although the number of determinations increased during the study period, the reason not to screen for AATD was not clear in many cases.11 Results further showed AATD determinations were highest for the 55–64 and 45–54-year old age groups.
A world map of the prevalence of the Pi*ZZ AATD genotype estimated there to be 20,611 Pi*ZZ individuals in Germany, 17,191 in France, 14,522 in Spain, 11,939 in England, 10,652 in Italy, 4,944 in Portugal, and 4,090 in Denmark.12 Focussing on the data from Spain, it is notable that only 500–600 individuals have been enrolled in the registry, representing just 3.6% of individuals with Pi*ZZ.
A recent ERS statement recommended that the management of AATD patients should be supervised by national or regional expert centres and that those centres should undertake systematic collection of clinical characteristics and disease natural history to enhance knowledge about the evolution and optimal management of AATD.4 The ERS statement additionally outlined criteria required for rare disease reference centres, including demonstration of a multidisciplinary approach and the ability to perform systematic collection of data in national and international registries. Registries are needed for orphan diseases to further increase knowledge about the disease and promote clinical research, gather epidemiological data, enable the collection of data for sufficiently powered clinical trials, promote collaborations between countries and researchers, and ensure long-term viability.
Since the Alpha-1 International Registry (AIR) was first established in 1997, it has facilitated collaborations between clinicians from 21 countries, integrated and promoted national registries, increased awareness of AATD in countries with low-to-medium prevalence, and enabled recruitment to two clinical trials. The annual accumulation of patients in the AIR registry rose from 519 in 1997 to 4,758 in 2012, when the collection of follow-up data stopped.13
As already mentioned in the previous presentation, central to the aims of EARCO, launched in 2017 by the ERS, is the establishment of the EARCO registry. Additional goals for EARCO include building a network of AATD researchers and clinical experts, reaching a consensus on the main research priorities, supporting and encouraging young investigators and clinicians, facilitating applications to industry and European Union (EU) funding sources, and starting standard operating procedures for sample processing and handling.
The EARCO registry, planned for launch in 2019, aims to recruit 3,000 patients with AATD over the first 3 years and gather information from centres across Europe. Data will be integrated with information from pre-existing national registries, allowing registries to grow together. Additional goals include facilitating development of longitudinal follow up, collaborations with other registries (such as the liver registry), and attracting new researchers and clinicians to AATD. These aims are reflected in the broad spectrum of data that will be collected, including sociodemographic and clinical data, with additional information on complementary tests, treatment, symptoms, and exacerbations. Patients will be followed up every 12 months, with emphasis placed on quality control and the uploading of spirometry and blood test results as PDF documents. Opportunities for recording data will be open to all physicians, with direct access from the EARCO website and links to national registries developed for sharing data. Economic incentives are planned to ensure the viability of the registry.
The case for a new AATD registry can be made to increase physicians’ awareness of and testing for AATD and enable recording of large-scale data that provides information beyond clinical trials. To conclude, the new EARCO registry will integrate existing national registries, enhance long-term follow-up and quality of data, and facilitate research and quality improvement initiatives across healthcare systems.
As a closing remark, Dr Barrecheguren reminded the audience that in a World Health Organization (WHO) meeting in 1997 recommended that COPD patients should be screened at least once for AAT deficiency14 and any patients showing abnormal screening results should undergo AAT protease inhibitor typing.
No Lab, No Diagnosis: How to Align Testing for Alpha-1 Antitrypsin Deficiency Across Europe
Doctor Ilaria Ferrarotti
The European Reference Networks are virtual networks involving healthcare providers across Europe and support patients with rare, low-prevalence, and complex diseases. One of the goals of the ERN-LUNG AATD Core Network is to establish a cross-border patient blood sample exchange to detect rare AAT gene mutants and create a quality validation programme across Europe to assess the quality and standardisation of laboratories inside the network. The plan is to establish a network of certified laboratories with AATD Core Network members in Warsaw and Pavia as the reference laboratories.
Laboratory diagnosis of AATD involves both quantitative and qualitative testing. Quantitative testing (evaluating AAT blood concentrations) can take place in any laboratory, while qualitative testing (aiming to correctly identify mutated alleles) can only take place in dedicated laboratories. Where expert laboratories are unavailable, AATD diagnosis cannot be considered reliable. AATD diagnostic laboratories offering state-of-the-art techniques and standard procedures performed by well-trained and dedicated personnel have been shown to reduce costs.15,16
Many variations of testing algorithms are possible. Diagnostic AATD testing can be performed on samples including blood, serum, or dried blood spot. Quantitative testing, used to measure AAT concentrations and C-reactive protein, can be undertaken with nephelometry, turbidimetry, ELISA, and immunodiffusion. Qualitative testing includes genotyping (limited to S and Z alleles or addressed to other pathological alleles), phenotyping, sequencing involving only exons, and sequencing involving the whole gene or even next-generation sequencing. In the last 10 years, a number of different algorithms have been proposed for AATD diagnosis by different reference centres that relate to the requirements of different countries and AATD populations.15,17-22
Another factor is that cut-off levels for diagnostic AAT serum values have varied. Historically, the AAT serum level was considered to be 11 µM,23 but more recently levels have moved to between 1.1 g/L24 and 1.00 g/L.18 The definition of the appropriate cut-off value will have significant impact on the detection of heterozygotes and rare alleles. While around 95% of diagnosed patients with severe AATD carry the PI*ZZ genotype, little is known about the epidemiology of the remaining variants, which are called rare because of their low prevalence and include PI*I and PI*Mmalton.25 Clinicians should be aware that AATD is not limited to Z and S alleles and that rare variants are only apparent if properly investigated. For example, detection of rare variants in the Italian AATD registry increased from 2000– 2004 and 2015–2017 due to technological progress and their prevalence exceeds 10% of all individuals with severe AAT deficiency.26
In July 2004, a Laboratory Alpha 1-antitrypsin Project workshop for scientists involved in AAT testing held in Stresa, Italy, first considered how to ensure quality control. Quality control consists of different components: pre-analytic (including sample collection, transport, and receipt), analytic (testing), and post-analytic (record keeping and reporting).
The European LAB-NET for AATD has been developed with the objective of introducing a pilot quality control programmes for six leading European laboratories involved in AATD diagnosis. The accuracy of diagnostic procedures will be verified at the laboratories in Marburg, Warsaw, Pavia, Barcelona, Lille, and Dublin, with Pavia and Warsaw providing the reference laboratories for shipping material. The initiative, which started in 2018, has been endorsed by the European Reference Networks and ERS, and is sponsored by CSL Behring.
Alternative Dosing Regimens for Patients with Alpha-1 Antitrypsin Deficiency: Safety of Bi-Weekly Alpha-1 Antitrypsin Therapy
Doctor Tim Greulich
While the recommended and licensed dose of AAT is 60 mg/kg body weight per week,4 the clinical reality is that weekly trips to hospitals for augmentation therapy infusions often prove unfeasible for patients. Taking a pragmatic approach, the Spanish Society of Pulmonology and Thoracic Surgery (SEPAR) guidelines for the management of AATD recommend alternative augmentation therapy dosing regimens, including 120 mg/kg for 14 days, 180 mg/kg for 21 days, and 50 mg/kg for 7 days.27 Such guidelines raise questions as to whether such alternative dosing regimens maintain sufficient AAT levels. The trough levels, i.e., levels just before the next infusion, should be greater than 0.5 g/L, which is equivalent to 11 µM.
A study evaluating whether steady state minimum concentrations of total AAT can be maintained above the threshold of 0.5 g/L with longer intervals between doses showed that both 100 mg/kg and 120 mg/kg every 14 days maintained serum trough levels >0.5 g/L but that serum trough levels were <0.5 g/L for 150 mg/kg and 180 mg/kg every 21 days and for 250 mg/kg every 28 days.28 In a Danish trial, where AATD patients were randomised to either AAT 250 mg/kg or placebo albumin 625 mg/kg infusions every 4 weeks, levels of AAT in the active arm were maintained above the 11 µM threshold for an average of 23–24 days, with a mean AAT level of 8.8 µM after 28 days.29 A simulation model of serum levels undertaken for the RAPID programme showed that for 60 mg/kg weekly the Cavg was 21.9 µM and the Ctrough was 16.3 µM; for 120 mg/g bi-weekly the Cavg was 21.9 µM and the Ctrough was 12.8 µM.30 The mean plasma concentrations were similar for both regimens and trough levels were maintained above the 11 µM threshold. Data from population pharmacokinetic simulations predicted that trough levels above the 11 µM protective threshold would be maintained in the majority of patients treated with bi-weekly 120 mg/kg infusions.30,31 (CSL Behring, data on file). Furthermore, a pharmacokinetic model suggested a linear relationship between AAT dose and AAT serum levels within the concentrations investigated.31
While a number of studies suggest that sufficient trough levels can be achieved with AAT 60 mg/kg every 7 days and 120 mg/kg every 14 days, little information has been published on the safety of bi-weekly infusions of 120 mg/kg AAT.
In the RAPID-RCT,32 where 180 AATD adult patients were randomised to AAT (n=93) or placebo (n=87), bi-weekly infusions were allowed under certain circumstances, for example, covering holiday periods. RAPID-RCT was followed by the RAPID- OLE study,33 where patients originally prescribed placebo could change to augmentation therapy.
In a post-hoc pooled analysis, the safety and tolerability of bi-weekly infusions of 120 mg/kg AAT were compared with weekly 60 mg/kg infusions for patients in both RAPID-RCT and RAPID-OLE.34 For the analysis, TEAE were defined as an AE not present before initiation of treatment or any AE that worsened in severity following exposure to treatment. Two different measures comparing AE rates were calculated: exposure-adjusted event rates (EAER), defined as all TEAE occurring during the trial divided by the cumulative exposure of the respective group (60 or 120 mg/kg AAT or matching placebo), and infusion-adjusted event rates as TEAE occurring in the 7-day period up to and following a bi-weekly 120 mg/kg infusion or matching placebo (Table 1).35
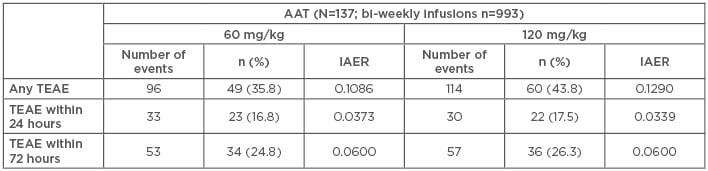
Table 1: Pooled data from the RAPID randomised control trial and the open label extension trials- comparing differences in treatment emergent adverse events between weekly AAT (60 mg/kg) and bi-weekly AAT (120 mg/kg).35
IAER: infusion-adjusted event rates; TEAE: treatment emergent adverse events.
During RAPID-RCT, 75 patients in the AAT group received 333 infusions with 120 mg/kg AAT during 321 sequences and in the pooled dataset of RAPID-RCT and RAPID-OLE studies, 933 bi-weekly infusions during 884 sequences in 137 patients were identified. Results show that the EAER was 5.8844 for patients receiving 60 mg/kg versus 5.7038 for patients receiving 120 mg/kg (p=0.681); for serious TEAE, the EAER was 0.3741 for 60 mg/kg versus 0.4074 for 120 mg/kg (p=0.850). Furthermore, the infusion-adjusted event rates for any TEAE were 0.1041 for 60 mg/kg versus 0.1267 for 120 mg/kg (p=0.3449); for serious TEAE, they were 0.0057 for 60 mg/kg versus 0.0079 for 120 mg/kg (p=1.0). The study showed that TEAE occurring within the first 24 and 72 hours after infusion were comparable between the subgroups. A breakdown of severity of reported TEAE showed that the majority of TEAE were mild or moderate and comparable between the 60 and 120 mg/kg groups.34
In conclusion, a post-hoc analysis of the RAPID programme showed that bi-weekly dosing with 120 mg/kg AAT had similar rates and severity of AE to weekly dosing with 60 mg/kg AAT.
Self-Administration of Alpha-1 Antitrypsin Therapy in the USA: Results of an Alphanet Survey
Professor Robert Sandhaus
AlphaNet is a not-for-profit American organisation that provides customised care and innovative disease management for AATD patients. In addition to funding AATD research, AlphaNet provides a comprehensive and structured disease management programme (ADMAPP) designed to optimise AATD outcomes. The overall goal is to reduce exacerbations, improve quality of life, and manage health resources efficiently.
ADMAPP is linked to a prospective outcome study involving >6,000 patients receiving AAT therapy. Currently, 65 regional AlphaNet co-ordinators (who are also AATD patients) provide regular contact with patients, and monthly questionnaires are used to collect data on well-being and care utilisation.
Medical interest in self-infusion was first triggered when it became apparent that patients who needed to travel for work had been teaching themselves to self-infuse. Self-administered home intravenous therapy is already common practice for hereditary angioedema, where it has been shown to deliver a range of benefits including enabling patients to resume normal work and family life (including travel), play an active role in disease management, and experience fewer healthcare visits and fewer episodes of hospitalisation.36
AlphaNet has recently undertaken a cross-sectional, observational telephone survey of ADMAPP patients to assess the occurrence of AAT self-administration. To take part, patients needed to be aged >18 years, diagnosed with AATD, and receiving AAT therapy with a human alpha 1-proteinase inhibitor (Zemaira® [CLS Behring, King of Prussia, Pennsylvania, USA] or Respreeza® [CSL Behring UK Ltd., Haywards Heath, UK).37 Altogether, 555 AATD patients answered the questionnaire, of which 170 (30.0%) were men and 214 (38.6%) women; the remainder (30.8%) did not disclose their sex. The age range of survey participants was 0.9% <30 years, 1.6% 30–39, 7.7% 40–49, 23.6% 50–59, 25.9% 60–69, 10.5% 70–79, and 1.4% >80. The occupation of respondents was students (1.1%), working full-time (14.4%), working part-time (8.5%), and retired (53.7%), with no response concerning employment from 22.3% of respondents.37
Results showed that 7.9% of respondents self-administered AAT versus 92.1% who did not. Of respondents who did not self-administer at the time of the survey, 2% reported that they had previously done so, with 30% of these patients reporting that the reason they gave up was due to inability to self-administer independently.37
From the survey, the most frequently cited reason for not considering self-administration was satisfaction with an existing regimen (79.9%). Other reasons cited included lack of confidence (26.2%), physical impairment (3.8%), and fear of needles (5%).37
Of the 44 patients who self-administered AAT, 95.4% reported being very satisfied and 4.6% were satisfied. The questionnaire also revealed that significantly more women than men self-administered treatment (p=0.007). Concerning how patients first heard about self-administration, 39.5% learned from other AATD patients, 14.0% from a home or specialist nurse, 7% from a nurse, and 4.7% from a doctor.37
The survey showed self-administration training was provided by home nursing agencies (72.1%), patients being self-taught (9.3%), training from hospital nurses (7.0%), and doctors (4.7%). Additionally, 6.9% of self-administering patients had received no training.37 Of patients who self-administered, 56.4% had received on average 2–3 training sessions. Most respondents (93%) cited greater independence as the biggest benefit of self-administration, with 37.2% saying it allows travel, 32.6% fits their lifestyle better, 2.3% saves time, 2.3% allows more control, 2.3% lowers medical costs, and 2.3% lowers transportation costs.37
Overall, 83.7% of patients who self-administered AAT therapy reported that they had experienced no difficulties with self-administration; of those experiencing difficulties, 5 reported infusion-related issues, including selection of infusion sites, ports becoming clogged with scar tissue, intravenous stick injuries, difficulties locating veins, and switching from vein to port.37
An unadjusted comparison of survival data from the ADMAPP programme with published historical data seems to imply that AATD patient outcomes have improved over the past 2–3 decades with the advent of augmentation therapy and ADMAPP and further analyses are ongoing to substantiate this finding and calculate the extent of this effect.
To conclude, the survey showed that patients who self-administer AAT therapy are very satisfied, have improved independence, and require limited training to self-administer independently.






