Abstract
Biosimilars are more affordable versions of previously approved biopharmaceuticals that are designed to reduce healthcare expenditure and increase patient access to this therapeutic class. To achieve their economic potential, many European countries have started to switch patients from reference drugs to biosimilars. The purpose of this article is to provide a comprehensive perspective on the biosimilar switching controversy, to assess interchangeability regulation and switching policies, and to review current evidence on switching and immunogenicity in the context of inflammatory rheumatic conditions. Patients and physicians feel uncertain about switching highly complex and difficult-to-replicate biosimilars of monoclonal antibodies due to a theoretical risk of increased immunogenicity, especially in extrapolated indications and in a multiple switch scenario involving various biosimilars. However, past experience with smaller biosimilars (somatropin, filgrastim, epoetin), the high standards required for approval of biosimilars of monoclonal antibodies in the European market, and current evidence on switching to infliximab and etanercept biosimilars (especially CT-P13 and SB4) are reassuring. Furthermore, no increased immunogenicity has been reported after switching to biosimilars. Decisions on switching and interchangeability are not covered by the European Medical Agency (EMA) guidelines and are left to individual European states, as opposed to the U.S. Food and Drug Administration (FDA), which has set standards to assess interchangeability. In summary, current knowledge is in favour of switching to biosimilars but the authors consider that this should be a physician-led decision with the active contribution of patients and hospital pharmacists to the pharmacovigilance chain.
INTRODUCTION
Biosimilars are similar and more affordable versions of previously approved biopharmaceuticals entering the market after loss of patent exclusivity. They present no clinical benefit over the originators and their use is aimed at reducing healthcare expenditure and improvement of patient access. In Europe, they are expected to mitigate access inequities between Eastern, Northern, and Western countries, the former having fewer reimbursed biologicals and prices that far exceeded the countries’ gross domestic product (GDP).1 The first biosimilar for the treatment of inflammatory conditions was approved in 2013 by the European Medicines Agency (EMA)2 and since then others have followed. However, biosimilar uptake has been slow and heterogeneous among European countries.3 Drivers for penetration of biosimilars include market dynamics, incentive policies (such as quotas), and price discounts. One important driver is non-medical switching from a reference drug to a biosimilar, determined by country-level policies. A non-medical switch occurs when a biopharmaceutical is replaced by another for reasons not related to efficacy or safety (usually economic). To fully achieve the cost-saving potential of biosimilars, many European countries have started switching patients to biosimilar drugs.
This article will explore the reasons behind biosimilar switching controversies, as well as review regulations on interchangeability, current switching data, and immunogenicity in the treatment of inflammatory rheumatic diseases.
WHY IS BIOSIMILAR SWITCHING AN ISSUE?
Switching from a reference biopharmaceutical to a biosimilar in a patient with an inflammatory condition is still a matter of debate. Biotechnological drugs are generated from living organisms and have inherently variable high-order structures (secondary, tertiary, and quaternary folding) and post-translational modifications (such as glycosylation, disulphide bond formation, or amidation) that impact structure, function, and immunogenicity. For these reasons, it is not possible to replicate a biopharmaceutical as an exact copy of the reference product, rendering biosimilars similar but not identical to their originators.4 Biosimilar manufacturers are required to follow regulatory standards to ensure that this expected variability remains within prespecified ranges.5 Developing a biosimilar candidate is both complex and laborious and typically involves characterising critical quality attributes and reverse-engineer manufacturing of reference product (cell culture, upstream, harvest, and downstream processes). Each one of these steps may introduce unwanted variability, and therefore manufacturers must apply state-of-the-art bioanalytical assays and confirmatory clinical trials to ensure maximal similarity of the end product.6
Biosimilars were first introduced in the European market following approval of Omnitrope® (Sandoz, Holzkirchen, Germany), the biosimilar of somatropin (human growth hormone), in 2006. Until 2013, all licensed biosimilars were either hormone (somatropin) or glycoprotein (filgrastim, epoetin alfa, and zeta) analogues.7 The first biosimilar of the monoclonal antibody infliximab was granted marketing authorisation in 20132 and, since then, biosimilars of etanercept, rituximab, adalimumab, and new biosimilars of infliximab were approved.7
Prospective and retrospective data have shown no significant safety or efficacy discrepancies following switch from reference to biosimilar hormones or glycoproteins. Somatropin, for instance, has the longest post-approval period and substantial cumulative data that revealed no unexpected adverse events and sustained efficacy in extrapolated indications and after switch.8,9 Filgrastim and complex glycoproteins, like epoetin alfa and epoetin zeta, have shorter post-approval periods but larger numbers of treated patients, and no difference in relevant clinical outcomes after switch.10,11
Notwithstanding this favourable historical background, switching biosimilars in the context of chronic inflammatory conditions has found resistance among patients and physicians due to concerns attributable mostly to immunogenicity.12-14 The rationale is that monoclonal antibodies and fusion proteins are much more difficult to replicate and may be more susceptible to immunogenic reactions. They have incommensurably more complex high-order structures and post-translational modifications, reaching close to 150 kDa of molecular weight compared to 30–40 kDa of hormones or glycoproteins.6 Furthermore, immunogenicity may be elicited not only from protein structure and post-translational modifications, but may also be process-related (impurities, aggregates, formulation, and storage conditions).
An immunogenic reaction characterised by anti-drug antibody (ADA) production is expected when two antigenically distinct proteins are switched. By definition, biosimilars must be antigenically similar to their originators. As depicted later in this paper, the majority of approved biosimilars in regulated markets have pre and post-approval studies confirming no increased immunogenicity after one or just a few switches, performed in monitored clinical settings; however, a scenario not tested is multiple switches between biosimilars. All biosimilars are tested against their reference product and may have minor differences in physicochemical or biological properties that have no impact on efficacy, safety, or immunogenicity. In upcoming years, there will be various biosimilar versions of the same reference product in the market and these may be used interchangeably as instructed by government authorities or hospital administrations. Considering that biosimilars are not required to demonstrate similarity amongst themselves and that numerous manufacturing changes occur throughout their life cycle, there is a theoretical risk that two biosimilars of the same reference product may diverge and become molecules with significant structural variations.15 Repeated exposure to such molecules with different stabilities or aggregation behaviour may increase the risk of immunogenic reactions with deleterious consequences for safety and efficacy.
Another concern, aside from immunogenicity, relates to pharmacokinetics (PK) and pharmacodynamics. All approved biosimilars have demonstrated a similar PK and, when available and relevant, pharmacodynamic profiles of their reference drug in a Phase I clinical trial.5 This is particularly important for large proteins such as monoclonal antibodies that may have variable PK behaviour even within the same disease population.4 When we consider scenarios that are not contemplated during the clinical assessment of a biosimilar candidate, patients and physicians feel uncertain. In a real-life setting, for instance, in which patients have several comorbidities and are treated with multiple concurrent drugs, there is a theoretical but remote risk that a biosimilar may behave differently from its reference drug, especially considering a disease condition for which no clinical studies were performed (extrapolated indication) and a multiple switch scenario.
REGULATION ON INTERCHANGEABILITY AND BIOSIMILAR SWITCHING POLICIES
The only regulatory agency with available guidance on interchangeability is the U.S. Food and Drug Administration (FDA).16 The Biologics Price Competition and Innovation Act (BPCIA) of 2009 distinguishes biosimilarity from interchangeability, stating that an interchangeable product must prove biosimilarity but is required to undergo further testing to demonstrate no risk to safety or efficacy of switching back and forth with the reference product.17 To comply with this legal requirement, the FDA published ‘Considerations in Demonstrating Interchangeability With a Reference Product – Guidance for Industry’16 in January 2017 so that manufacturers could apply and have their biosimilars additionally licenced as interchangeable. This extensive draft guidance provides an overview on scientific considerations in demonstrating interchangeability, including data and information needed to support a demonstration of interchangeability; design and analysis of a switching study or studies; recommendations regarding the use of a USA-licensed reference product in a switching study or studies; and considerations for developing presentations, container closure systems, and delivery device constituent parts for proposed interchangeable products.16 Furthermore, the BPCIA stated that once a biosimilar is licenced as interchangeable, pharmacy-level substitution may occur, meaning that a reference biopharmaceutical may be substituted at the pharmacy to the interchangeable version without the prescriber’s consent.17 The additional amount of data required to apply for a licence as an interchangeable product adds further costs to the development programme of a biosimilar. However, this investment is likely to provide return as it opens the door for automatic substitution and bypasses physician and patient resistance to switching. The first biosimilars approved as interchangeable are expected in the USA market in late 2018 or early 2019.
The EMA has been at the frontline of biosimilar regulation, issuing the first overarching guideline in 2005 and many other product-specific recommendations since then. However, interchangeability is not covered in the EMA guidelines and the decisions on interchanging and substituting are left to individual member states, which have access to the scientific evaluations performed by EMA’s committees.18 As a consequence, the European reality on this matter is somewhat heterogeneous. Scandinavian countries, such as Norway and Denmark, featured among the first to adopt an administrative-driven, large-scale switch from reference infliximab and etanercept to their corresponding biosimilars. National regulatory agencies from other countries, including France, England, the Netherlands, and Portugal, have recommended the adoption of switching policies and the transition to infliximab and etanercept biosimilars is starting to occur.19-21
In Europe, biosimilar acceptance has grown among patients and physicians despite the lack of structured educational programmes in most of these countries. Nonetheless, national rheumatology societies and patient associations have expressed their concerns on non-medical switching and interchangeability. Table 1 presents the position statements of rheumatology societies from European countries.22-29 In summary, automatic substitution is consensually rejected because physicians consider it a risk to traceability and pharmacovigilance. Some societies are starting to accept non-medical switching if the physician remains at the centre of the switching process and certain conditions are met. Interchangeability is currently not recommended by most due to the limited evidence on multiple switching.
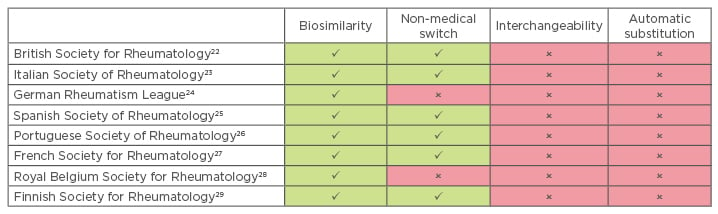
Table 1: Position of rheumatology societies from European countries on biosimilar switching, interchangeability, and automatic substitution.
Green: acceptance; red: non-acceptance.
CURRENT EVIDENCE ON BIOSIMILAR SWITCHING
Not surprisingly, the greatest amount of data on biosimilar switching in the context of inflammatory rheumatic conditions comes from CT-P13 (Remsima®, Celltrion, Incheon, South Korea; Inflectra®, Hospira, Lake Forest, Illinois, USA), the biosimilar of infliximab, which was the first monoclonal antibody approved, almost 5 years ago. Nevertheless, it is important to note that these data comprise almost exclusively open-label extensions of randomised double-blind trials30,31 and registry or single-centre observational studies, they assess only one transition from reference drug to biosimilar, and many lack appropriate control arms. One exception is the NOR-SWITCH trial,32 a double-blind Phase IV trial in which 482 patients (inflammatory bowel disease, axial spondyloarthritis [axSpA], rheumatoid arthritis [RA], psoriatic arthritis [PsA], and plaque psoriasis) from Norway on stable treatment with reference infliximab were randomised to continue on reference infliximab or switch to CT-P13. Disease worsening (primary endpoint) and safety at 52 weeks were not different between study arms in the overall population (95% confidence interval of group difference: -12.7–3.9%; 15% non-inferiority margin for disease worsening in the entire population), though this study was not powered to detect differences in individual disease groups.32 A nationwide, prospective, observational study from the DANBIO registry assessed switching from reference infliximab to CT-P13 in 802 patients with RA, axSpA, and PsA, which found no difference in disease activity 3 months before and after switching in each disease subset.33 One-year adjusted absolute retention rates but not crude retention rates were slightly lower compared to historical infliximab cohorts (83.4% versus 86.8%, p=0.03), which was attributed by the authors to probable nocebo effect and residual confounding.33 In line with the latter finding, Tweehuysen et al.34 concluded that subjective features were the main driver for discontinuation after 6 months of transition to CT-P13 in RA, axSpA, and PsA patients, also due to the probable nocebo-effect and incorrect causal attribution effects.34 Many other studies assessed open-label single transitions to CT-P13 in individual centres with a variable number of patients, but with overall positive results. Although inflammatory bowel disease is out of the scope of this article, it is noteworthy that a growing body of evidence supports that efficacy, safety, and immunogenicity remain unchanged after switching to CT-P13, including in the paediatric setting.35,36 Infliximab biosimilar SB2 (Flixabi®, Biogen, Cambridge, Massachusetts, USA) demonstrated comparable efficacy, safety, and immunogenicity to the reference drug in the extension of the Phase III trial in which RA patients receiving SB2 continued to receive SB2 and those receiving reference infliximab were re-randomised to either switch to SB2 or to continue on reference infliximab, from Week 54 to Week 78. This transition study maintained double-blind status and allowed for simultaneous comparison of the switched group with the ongoing reference and biosimilar groups.37
Evidence on switching to etanercept biosimilars is growing. The Phase III trial of etanercept biosimilar GP2015 (Erelzi®, Sandoz) was performed in a non-rheumatic population but is worth mentioning for its unique design of multiple-switching.38 Following the initial 12-week parallel-group period, patients with moderate-to-severe chronic plaque-type psoriasis either remained on the original allocated drug or interchanged treatment drug three times over 6-week intervals. After 52 weeks, the multiple-switch arms showed no efficacy, safety, or immunogenicity differences as compared to the maintenance arms.38 Full-text manuscripts assessing the switch to SB4 (Benepali®, Biogen, USA) include a Phase I single-blind PK study in healthy individuals and an open-label extension of the Phase III trial evaluating transition to SB4 up to Week 100 in RA patients, and neither reported any discrepancies in efficacy or safety outcomes after switch.39,40 Data from 1,623 RA, axSpA, and PsA patients from the DANBIO registry were presented as an abstract and revealed no significant change in disease activity 3 months after the switch to SB4; 9% (129) stopped treatment after 5 months follow-up largely due to lack of effect and adverse events.41 One-year results of this observational study were later presented as another abstract showing 18% (276 of 1,623 patients) treatment withdrawal but no update on efficacy outcomes was made.42 The BIO-SPAN study43 evaluated non-mandatory transitioning to SB4 in 635 RA, AxSpA, and PsA patients using a specific communication strategy to counter nocebo and attribution. Compared to baseline, there was no difference at 6 months in efficacy but persistence and decreases in DAS28-CRP and CRP were slightly lower for SB4 compared to an historical 2014 etanercept cohort.43
Adalimumab biosimilars have recently been approved and are expected to enter the European market in late 2018. Thus, evidence on switching is still scarce and is published mostly as abstracts. One exception is SB5 (Imraldi®, Biogen), for which there was a published double-blind Phase III trial demonstrating similar efficacy, safety, immunogenicity, and radiographic outcomes at 52 weeks in RA patients who switched from reference adalimumab to SB5 at Week 24 compared to maintenance arms.44 The BI 695501 (Cyltezo®, Boehringer Ingelheim, Germany) Phase III extension trial showed that a single transition had no impact on efficacy, safety, and immunogenicity in RA patients at 58 weeks when compared to those continuing on reference drug or BI 695501.45 Adalimumab biosimilar ABP 501 (Amgevita®, Solymbic®, Amgem, USA) has interim results from one open-label single-arm extension study in which the transition from the reference drug at Week 26 was associated with sustained efficacy and safety in RA patients at Week 72.46
Evidence on switching to rituximab biosimilars GP2013 (Riximyo® and Rixathon®, Sandoz, Germany) and CT-P10 (Truxima®, Blitzima®, Ritemvia®, Rituzena®, Celltrion, South Korea) in rheumatic conditions is still restricted to small-sized studies with limited reporting of efficacy and safety outcomes.47-49
It is noteworthy that evidence on switching these and other biosimilars is expected to grow in the near future because there are several ongoing studies in rheumatic and non-rheumatic inflammatory conditions.
SWITCHING AND IMMUNOGENICITY
Apprehensions have been raised that switching patients from reference antibodies to biosimilars may lead to increased immunogenicity and consequent safety or efficacy problems. Switches occur when patients receive biosimilars but may also occur after manufacturing process changes lead to structural modifications or changes in the impurity profile of the biologic drug.11,50 This situation occurred with multiple medicines such as darbepoetin or infliximab.51,52 A commonly expressed concern is whether there is an increase in immunogenicity related to the act of switching itself. ADA assays offer the most sensitive method to detect immunogenicity; neutralising antibodies (NAB) assays are the most direct method to signal the potential clinical relevance of ADA. PK, efficacy, and safety events may be additional measures to detect clinically relevant immunogenicity.4
The authors searched immunogenicity data from confirmatory trials of approved biosimilars in rheumatic diseases. Data collected included the proportion of patients positive for ADA among all patients and the proportion of patients with NAB among ADA-positive patients. The authors identified 10 biosimilars approved by the EMA or FDA: three for adalimumab (BI 695501, SB5, and ABP 501) and infliximab (SB2, CT-P13, and infliximab-qbtx), and two for etanercept (GP2015 and SB4) and rituximab (CT-P10 and GP2013). Published data in EMA Public Assessment Reports (EPAR), FDA Clinical Summaries, PubMed, and European League Against Rheumatism (EULAR) and American College of Rheumatology (ACR) abstracts show that the duration of treatment in the 16 identified trials (which varied in design and methodology of ADA and NAB detection) ranged from 12 to 102 weeks.38,53-57 The lowest proportions of ADA-positive (0–13%) and NAB-positive (0–3%) patients were observed in the trials of etanercept and its biosimilars, and the highest in the trials of infliximab and its biosimilars (ADA: 20–62%; NAB: 88–100%).38,53-57 The proportions of ADA and NAB-positive patients in individual trials were similar between the originator and biosimilar products. Of note, in a 52-week trial of etanercept biosimilar SB4, the incidence of ADA by Week 52 was significantly lower in the SB4 arm (1% [3/299] versus 13% [39/296]; p<0.001).53 This difference may have been due to an ADA assay bias in samples collected at Weeks 4 and 8. However, it was recently confirmed that SB4 has equivalent efficacy to reference etanercept but is associated with fewer injection site reactions and less immunogenicity. Clinical features were generally comparable between the treatment groups regardless of ADA status.58 Cross-reactivity between ADA of biosimilar and reference drugs suggests that epitopes influencing the immune response are common to both drugs.59-61
The results from immunogenic response to biosimilars in naïve patients are reflected in nearly all published studies evaluating switching between a biologic and a biosimilar. A recent study examining data from published literature showed no differences in immunogenicity, safety, or efficacy. This assessment covered seven molecular entities and 14,225 individuals from multiple indications between 1993 and June 2017, but a subset analysis of anti-TNF and anti-CD20 biosimilars demonstrates equivalent results.62 While there are limitations to some of the individual studies, the cumulative results of these published data do not show significant differences in ADA or NAB after switching compared to subjects who were not switched. There was also no reported increase in treatment-related safety events, including loss of efficacy. Only two studies report loss of efficacy or high dropout rates after switching from reference medicine to biosimilar infliximab; the results of Kang et al.63 and Yazici et al.64 studies were not replicated in other studies of switching from reference to biosimilar infliximab. Although most studies evaluate the effects of a single switch, the authors argue that long-term experience with biologics (including interchanging between biologicals and between pre and post-modification batches of the same drug) gives a strong indication that multiple switches would not create problems for patients. However, further studies are warranted to confirm this hypothesis.
CONCLUSION
Current knowledge is favourable to switching from reference drugs to biosimilars in the treatment of inflammatory rheumatic conditions. However, one must consider that evidence comes essentially from a few observational studies and double-blind or open-label extensions of Phase III trials, performed on a reduced number of patients with limited duration of follow-up, mostly on CT-P13 and SB4. This evidence cannot be extrapolated to other biosimilars and it is arguable whether it should be extrapolated to other conditions for which the biosimilar is approved. There will always be a knowledge gap because studies do not cover all the switching possibilities taking place in real life. It is highly unlikely that manufacturers hold trials assessing switch between different biosimilars because this would represent additional costs and still provide insufficient answers.
It is the authors’ strong belief that a robust state-of-the-art demonstration of biosimilarity combined with rigorous post-marketing pharmacovigilance mechanisms involving pharmacists, prescribers, and patients will bring reassurance to switching and interchangeability. Prescribers and pharmacists should ensure adequate registration of biosimilar trade name and batch number. Physicians should be encouraged to spontaneously report adverse events and use national registries to document efficacy, safety, and immunogenicity after switching. Patients should be fully knowledgeable about the biopharmaceutical they were prescribed and properly educated on how to report possible adverse events.
It is also the authors’ belief that the prescribing physician should be in the centre of the switching decision. This decision should be made on a case-by-case basis taking into consideration patient and disease characteristics, as well as drug and device-related factors. National or regional authorities may compel hospital pharmacies to automatically substitute a reference biologic for a biosimilar as a means to rapidly achieve cost containment. For the time being, we consider this administrative substitution unacceptable because it compromises the chain of pharmacovigilance, and ultimately endangers not only the safety of patients but also the future of biosimilars in the treatment of rheumatic conditions.