This is a summary of selected data presented at the 15th Georg Rajka International Symposium on Atopic Dermatitis (ISAD), organised by the International Society of Atopic Dermatitis, held in Melbourne, Australia, from 24th–26th October, 2025; the 45th Annual Fall Clinical Dermatology Congress, held in Las Vegas, Nevada, USA, from 23rd–26th October, 2025; and the European Academy of Dermatology and Venereology Congress 2025, held in Paris, France, from 17th–20th September, 2025
Support: The development and publication of this content was fully funded by LEO Pharma A/S and is intended for healthcare professionals only.
Presenters: Andreas Wollenberg,1 Teodora Festini,2 Ahmed Ameen,3 April W. Armstrong,4 Diamant Thaçi5
1. Department of Dermatology and Allergy, Augsburg University Hospital, Germany
2. LEO Pharma A/S, Ballerup, Denmark
3. David Geffen School of Medicine, University of California Los Angeles, USA
4. Division of Dermatology, Department of Medicine, David Geffen School of Medicine, University of California Los Angeles, USA
5. Institute and Comprehensive Centre for Inflammation Medicine, University of Lübeck, Germany
Disclaimer: The opinions expressed in this article belong solely to the authors of the posters. For UK and Ireland Healthcare professionals, UK prescribing information for Adtralza®▼ (tralokinumab) can be found here.▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See Section 4.8 for how to report adverse reactions. For the Republic of Ireland, the abbreviated prescribing information can be found at the end of this article. Adverse Event reporting information for the UK and Ireland can be found at the end of this article.
UK summary of product characteristics for the 150 mg solution for injection in pre-filled syringe can be found here.
UK summary of product characteristics for the 300 mg solution for injection in a pre-filled pen can be found here.
EU summary of product characteristics can be found here.
Tralokinumab is indicated for the treatment of moderate‑to‑severe atopic dermatitis in adult and adolescent patients 12 years and older who are candidates for systemic therapy.
Acknowledgements: Writing assistance was provided by Christos Evangelou, San Diego, California, USA.
Disclosure: Wollenberg has served as an advisor or paid speaker for, or participated in clinical trials (with honoraria paid to the institution) sponsored by AbbVie, Aileens, Almirall, Amgen, Apogee, Beiersdorf, Bioderma, Bioproject, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Chugai, DKSH, Eli Lilly, Galapagos, Galderma, Glenmark, GSK, Hans Karrer, Hexal, Incyte, Janssen-Cilag, Kyowa Kirin, LEO Pharma, L’Oreal, Maruho, MedImmune, MSD, Mylan, MSD, Nektar, Novartis, Pfizer, Pierre Fabre, Regeneron, Sandoz, Santen, Sanofi, and UCB. Festini is an employee of LEO Pharma A/S. Ameen has been a speaker, advisor, and/or investigator for AbbVie, Bayer, Boehringer Ingelheim, Ego Pharmaceuticals, Galderma, Jamjoom Pharma, Janssen, LEO Pharma A/S, Lilly, Novartis, Organon, Pfizer, Sanofi, and Viatris. Armstrong has served as a research investigator and/or scientific advisor for AbbVie, Almirall, Arcutis Biotherapeutics, ASLAN, Beiersdorf, Boehringer Ingelheim, Bristol-Myers Squibb, Dermavant, Dermira, EPI, Incyte Corporation, Janssen, LEO Pharma A/S, Lilly, Modmed, Nimbus, Novartis, Ortho Dermatologics, Pfizer, Regeneron, Sanofi, Sun Pharma, and UCB. Thaçi has served as an investigator and/or consultant/adviser for AbbVie, Almirall, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Celltrion, Eli Lilly, Galderma, Johnson & Johnson, LEO Pharma A/S, L’Oreal, Meiji, NewBridge Pharmaceuticals, Novartis, Pfizer, Regeneron, Sanofi, Target RWE, UCB, and Vichy; and has received grants from AbbVie and LEO Pharma A/S.
Keywords: Atopic dermatitis (AD), genital dermatitis, hand dermatitis, head and neck dermatitis, IL-13, tralokinumab.
Meeting Summary
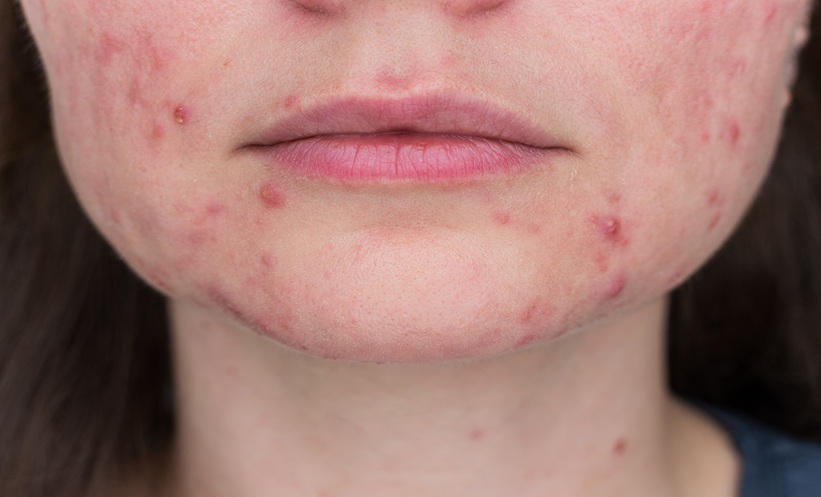
Atopic dermatitis (AD) affecting the head and neck, hands, and genitals is associated with a high burden of impact on quality of life. Tralokinumab has been approved in several jurisdictions, including in the European Economic Area, Ireland, and the UK, for the treatment of moderate-to severe AD in adult and adolescent patients 12 years and older who are candidates for systemic therapy. Clinical trial data and real-world evidence, presented at the Georg RAJKA International Symposium on Atopic Dermatitis (ISAD), indicate that tralokinumab may provide disease control in individuals with moderate-to-severe AD in high-burden areas. The Phase IIIb ADHAND trial investigated tralokinumab monotherapy in adults with AD and moderate-to-severe hand involvement who are candidates for systemic therapy. At Week 16, 40% of tralokinumab-treated patients had clear or almost clear hands, compared with 10.6% on placebo. The real-world TRACE study demonstrated that, among patients with AD with head and neck involvement at baseline, 64.7% (205/317) showed Investigator’s Global Assessment (IGA) 0/1 by Month 12, and among those with baseline genital involvement, 78.3% (54/69) experienced AD clearance by Month 12. The ECZTEND open-label extension study demonstrated that improvements in moderate-to-severe AD with head and neck involvement were maintained over 5 years. Across studies, the reported safety profile was consistent with the approved label. Safety analysis from TRACE revealed that among patients who discontinued dupilumab due to conjunctivitis, 82.1% (32/39) did not develop tralokinumab-related conjunctivitis. When conjunctivitis occurred during tralokinumab treatment, it was mild-to-moderate and did not lead to treatment discontinuation. These findings indicate that tralokinumab was associated with improvements in disease severity in patients with moderate-to-severe AD involving high‑burden areas, including individuals who had discontinued dupilumab because of conjunctivitis.Introduction
AD is a prevalent chronic inflammatory skin condition, affecting approximately 204 million individuals worldwide.1 Patients with AD often report lifestyle limitations, avoidance of social interaction, and impaired daily activities, and identify symptoms such as itch, excessive dryness or scaling, and red or inflamed skin as the most burdensome.2 Although AD can affect various parts of the skin, involvement of certain anatomical areas, including the head and neck, hands, and genital regions, is associated with particularly high disease burden in terms of extent of involvement and impact on quality of life.3,4
Head, face, and neck involvement occurs in approximately 72% (391/541) of patients with moderate-to-severe AD.3 Hand involvement affects 56% of patients with moderate or severe AD.3 Together, involvement of the head, face, neck, and hands is associated with higher median Dermatology Life Quality Index (DLQI) scores (a measure of how skin problems affect an adult patient’s life), higher rates of quality-of-life impacts, and greater psychological distress compared with AD affecting other body areas.3 Although underreported because of the patients’ reluctance to discuss this area, genital involvement can affect sexual function, reproductive desire, and overall quality of life.4,5
Tralokinumab is a fully human, high-affinity monoclonal antibody that binds IL-13, preventing receptor interaction and subsequent downstream signalling.6-8 It has been approved in several jurisdictions, including in the European Economic Area, Ireland, and the UK, for the treatment of moderate-to-severe AD in adult and adolescent patients 12 years and older who are candidates for systemic therapy.8-10 In the 52-week, randomised, multicentre, placebo-controlled Phase III ECZTRA 1 and ECZTRA 2 trials, tralokinumab monotherapy (300 mg subcutaneous every 2 weeks) improved IGA 0/1 rate compared with placebo (IGA 0/1 rate 15.8% [95/601] with tralokinumab versus 7.1% [14/197] with placebo in ECZTRA 1; 22.2% [131/591] with tralokinumab versus 10.9% [22/201] with placebo in ECZTRA 2).7 The rate of Eczema Area and Severity Index (EASI) responses (the proportions of patients achieving ≥75% improvement [EASI-75]) at Week 16 in patients with moderate-to-severe AD was 25.0% (150/601) with tralokinumab versus 12.7% (25/197) with placebo in ECZTRA 1, and 33.2% (196/591) with tralokinumab versus 11.4% (23/201) with placebo in ECZTRA 2.7 In addition, the randomised, controlled Phase III ECZTRA 3 trial demonstrated that treatment with tralokinumab 300 mg every 2 weeks plus topical corticosteroids improved IGA 0/1 (38.9% [98/252] with tralokinumab plus corticosteroids versus 26.2% [33/126] with placebo) and EASI-75 (56.0% [141/252] with tralokinumab plus corticosteroids versus 35.7% [45/126] with placebo) at Week 16 compared with placebo in adults with moderate-to-severe AD, with most patients showing sustained responses at Week 32.11
This article discusses recent data from a Phase IIIb clinical trial of tralokinumab in patients with AD with moderate-to-severe hand involvement who are candidates for systemic therapy, the TRACE real-world study on the efficacy of tralokinumab in the treatment of AD affecting the head and neck and genital regions, the role of tralokinumab in patients switching from other biologic therapies, and long-term data from an open-label extension trial.
Phase III Evidence from the ADHAND Trial
Study Design and Patient Characteristics
The ADHAND trial was a randomised, double-blind, placebo-controlled Phase IIIb study evaluating tralokinumab monotherapy in adults with AD and moderate-to-severe hand involvement who were candidates for systemic therapy.12 The study enrolled 235 patients who were randomised 2:1 to receive either tralokinumab 300 mg every 2 weeks (n=156) or placebo (n=79) for 16 weeks, followed by an open-label period in which all patients received tralokinumab for a further 16 weeks.12
The study population comprised adult patients with atopic hand eczema (AHE) with an IGA-AHE score of 3 or 4 (moderate-to-severe AHE), Hand Eczema Symptom Diary (HESD) itch score ≥4 at baseline, recent inadequate response to topical prescription medications, and AD involvement of at least one body location other than hands or wrists.12 Patients who received systemic medication within the prior 4 weeks or topical medication within the prior week of trial start, as well as those with prior failure on tralokinumab (lack of efficacy or safety concern) or dupilumab (lack of efficacy), were excluded.12
The study population had a median age of 37 years, and 63.4% of patients were female.12 Most patients were White (67%) or Asian (24%).12 The median duration of AD was 20 years, and the median age at onset of hand involvement was 24 years. All patients had received prior topical corticosteroid therapy, and 11.5% had previously received biologic therapy. At baseline, 27.7% of patients had severe AHE, the median Hand Eczema Severity Index (HECSI) was 68.0, the median HESD itch score was 7.57, and the median HESD pain score was 7.14.12
Improvement in Hand Involvement
The primary endpoint of the ADHAND trial was the proportion of patients with IGA-AHE 0 or 1 (clear or almost clear skin on hands) at Week 16.12 Tralokinumab significantly improved IGA-AHE 0/1 response rates compared with placebo (40.0% in the tralokinumab group versus 10.6% in the placebo group, representing a treatment difference of 29.7% [95% CI: 19.0–40.4; p<0.001]). Onset of effect was observed as early as Week 2 of treatment, and statistically significant differences in IGA-AHE 0/1 response rates between the tralokinumab and placebo groups were maintained throughout the 16-week double-blind treatment period.12
Improvements in Hand Eczema Severity Index Response
Secondary endpoints included various thresholds of HECSI improvement at Week 16.12 The proportion of patients with ≥75% improvement in HECSI (HECSI-75) at Week 16 was 64.1% in the tralokinumab group versus 33.8% in the placebo group, representing a treatment difference of 30.3% (95% CI: 17.5–43.0; p<0.0001). The more stringent HECSI-90 threshold was met by 41.7% of patients treated with tralokinumab, compared with 10.9% of those who received placebo, a difference of 30.8% (95% CI: 20.0–41.5; p<0.0001).12
Improvements inPatient-Reported Outcomes
Patient-reported outcomes showed greater improvements throughout the 16-week double-blind treatment period in the tralokinumab group compared with the placebo group.12 DLQI percentage change from baseline to Week 16 was −60.91% for tralokinumab versus −20.99% for placebo, representing a treatment difference of −39.92% (95% CI: −53.54–−26.31; p<0.0001). The percentage change in Hand Eczema Impact Scale (HEIS) from baseline to Week 16 was −64.89% for tralokinumab versus −28.55% for placebo, a difference of −36.34% (95% CI: −47.14–−25.55; p<0.0001).12
The percentage of patients with a ≥4-point reduction in HESD itch was 47.3% in the tralokinumab group and 20.7% in the placebo group (treatment difference: 26.6%; 95% CI: 14.6–38.7; p<0.0001).12 Furthermore, 45.3% of tralokinumab-treated patients reported a ≥4-point reduction in HESD pain, compared to 13.3% of placebo-treated patients (treatment difference: 32.0%; 95% CI: 20.8–43.1; p<0.0001).12 These results reflect outcomes observed in the study population and should be interpreted in the context of the overall evidence base.
Safety Profile
As with all clinical studies, efficacy outcomes should be interpreted alongside adverse event data. The safety profile of tralokinumab in the ADHAND trial was consistent with that in previous clinical trials, with no new safety signals identified throughout the 16-week treatment period.12 Adverse events occurred in 60.3% (94/156) of patients in the tralokinumab group and 60.8% (48/79) of those in the placebo group. Serious adverse events were reported in 1.9% (3/156) of tralokinumab-treated patients and in 1.3% (1/79) of placebo-treated patients.12
Conjunctivitis, a predefined adverse event of special interest, occurred in 3.8% of patients in both treatment groups.12 No patients discontinued treatment because of conjunctivitis or any other adverse event of special interest.12 The conjunctivitis rates observed in ADHAND were similar to those reported in previous Phase III studies of tralokinumab in moderate‑to‑severe AD.9,12-14
Real-World Evidence on Head and Neck Atopic Dermatitis: Insights from the TRACE Study
Patient Population and Baseline Characteristics
The TRACE study is an international, prospective, non-interventional study that followed 824 adults with AD who were prescribed tralokinumab in accordance with nationally approved labels for up to 12 months across 149 global sites.15
Among the TRACE study population, 79% (654/824 participants) reported head and neck involvement at baseline.15 Patients with head and neck involvement had a mean age of 42.1 years, and 53.2% were male. The population was predominantly White (77.2%), and the mean AD duration was 20.7 years. At baseline, 12.9% (68/525) of patients had an EASI score of ≤7, and 40% (248/619) had severe disease (IGA 4). The study population included both dupilumab-naive patients (n=510) and patients previously treated with dupilumab (n=144).15
Effectiveness in Head and Neck Atopic Dermatitis
The results demonstrated that improvements in physician-assessed effectiveness measures were maintained over the 12-month treatment period in patients with head and neck involvement.15 The proportion of participants with head and neck involvement decreased from 100% at baseline to 51.8% (37.6% were without head and neck involvement, and for 10.6% the outcome was unknown) at Month 12 in the total population (50.5% among dupilumab-naive patients [37.4% were without head and neck involvement, and for 12.1% the outcome was unknown] and 59.3% [38.9% were without head and neck involvement, and for 1.9% the outcome was unknown] among those who received prior treatment with dupilumab).15
Tralokinumab also improved composite effectiveness measures.15 Using a composite responder definition of achieving either IGA 0/1, EASI-75, or Scoring Atopic Dermatitis (SCORAD) <10, the response rate among patients with head and neck involvement at baseline increased from 5% (30/628) at baseline to 80% (263/329) by Month 12. A composite responder definition of achieving either IGA 0/1, EASI score ≤7, or SCORAD <10 showed similar trends, with the response rate increasing from 13% (82/651) at baseline to 87% (287/330) at Month 12.15
Changes in individual disease severity measures showed consistent improvements.15 The proportion of patients with IGA 0/1 increased from 1.4% (9/619) at baseline to 64.7% (205/371) at Month 12. EASI-75 response by Month 12 was reported in 79.2% (217/274) of patients, and EASI ≤7 was reported in 12.9% (68/525) at baseline, increasing to 89.3% (259/290) of patients at Month 12.15
Patient-Reported Outcomes
Mean DLQI scores decreased from 13.1 at baseline to 5.1 at Month 12.15 The proportion of patients achieving DLQI ≤5, indicating no to small quality of life impairment, increased from 19.0% (71/373) at baseline to 67.8% (97/143) at Month 12.15 Mean Peak Pruritus Numerical Rating Scale (PP-NRS) score, indicating itch severity, decreased from 6.4 at baseline to 3.3 at Month 12, and the percentage of patients with PP-NRS ≤4 increased from 21.0% (83/395) at baseline to 70.0% (98/140) at Month 12. Sleep disturbance also improved, with mean Sleep NRS decreasing from 5.2 at baseline to 2.2 at 12 months. Among participants with baseline Sleep NRS ≥2, 70.7% (58/82) reported a ≥2-point improvement by Month 12.15
Safety and Effectiveness in Patients Switching from Dupilumab Due to Conjunctivitis
Conjunctivitis can lead to permanent treatment discontinuation in some patients with AD who receive treatment with dupilumab.15-17 The TRACE study included 182 patients who received prior treatment with dupilumab, 39 (21.4%) of whom had discontinued dupilumab because of conjunctivitis.18 Patients who discontinued dupilumab because of conjunctivitis had a mean age of 48.0 years, and were predominantly male (56.4%) and White (69.2%).18
During tralokinumab therapy, 43.6% (17/39) of patients reported any adverse event, and 5.1% (2/39) discontinued tralokinumab due to an adverse event.18 Neither of these discontinuations was due to conjunctivitis.
Ten (25.6%) patients reported conjunctivitis during tralokinumab treatment, of whom seven (17.9%) had conjunctivitis assessed by investigators as possibly related to tralokinumab.18 Among the seven patients who experienced tralokinumab-related conjunctivitis, the severity was generally mild-to-moderate, with three cases classified as mild, three as moderate, and one as severe.18
In this 12‑month study, patients who switched from dupilumab to tralokinumab experienced numerical improvements in disease severity and patient‑reported outcomes over the treatment period.18 By Month 12, 81% (13/16) of patients had an EASI ≤7, compared with 44% (16/36) at baseline. Improvements were also observed in quality of life, with 83% (10/12) of patients achieving a DLQI ≤5 at Month 12 versus 54% (15/28) at baseline. Itch scores showed a similar pattern, with 78% (7/9) of patients reporting a PP‑NRS ≤4 at Month 12, compared with 59% (17/29) at baseline.18 These findings reflect outcomes in the study population and should be interpreted within the context of the study design and overall evidence base.
Real-World Evidence on Genital Involvement: Insights from the TRACE Study
Baseline Characteristics and Disease Severity
In the TRACE study, 15% of the total population (124 patients) had AD involving the genital area at baseline.19 Patients with genital involvement had similar baseline characteristics as those in the overall TRACE population, with some exceptions. They were more likely to be male (63.0% versus 52.4% in the total population) and White (81.5% versus 75.7%). In addition, patients with genital involvement had more severe disease at baseline, with mean EASI scores of 23.9, compared with 20.1 in the total population, and 49.2% (61/124) had severe disease by IGA compared with 34.1% (281/824) in the overall population. Among patients with genital involvement at baseline, mean DLQI was 15.7, mean Sleep NRS was 6.1, and mean PP-NRS was 6.9.19
Improvements in Genital Involvement and Disease Severity
By Month 12, 78.3% (54/69) of patients with baseline genital involvement no longer had genital AD.19 Genital involvement clearance occurred relatively fast, with 71.7% (76/106) of patients showing no genital involvement by Month 3, a proportion that remained high through Month 12. Using the composite definition of IGA 0/1, EASI-75, or SCORAD <10, response rates increased from 2% (3/122) at baseline to 79% (44/58) at Month 12. The alternative composite definition (IGA 0/1, EASI ≤7, or SCORAD <10) showed similar results, with the response rate increasing from 6% (7/124) at baseline to 80% (46/58) at Month 12.19
Individual components of composite measures of response demonstrated consistent improvements.19 At Month 12, 50.8% of patients had IGA 0/1, with an additional 30.5% of patients showing mild disease (IGA 2) by Month 12. At Month 12, EASI-75 response was reported in 76% (38/50) of patients, and EASI ≤7 was reported in 80% (40/50) of patients.19
Improvements in Patient-Reported Outcomes
Mean DLQI decreased from 15.7 at baseline to 7.0 at Month 12, and 59% (13/22) of patients had DLQI ≤5 by Month 12.19 Mean PP-NRS decreased from 6.9 at baseline to 3.9 at Month 12, and 56% (10/18) of patients had PP-NRS ≤4 by Month 12. Sleep quality also improved, with mean Sleep NRS decreasing from 6.1 at baseline to 3.4 at Month 12 and 63% (10/16) of patients reporting a Sleep NRS ≥2 improvement by Month 12.19
Long-Term Clinical Trial Data: 5-Year Results from ECZTEND
Study Design and Patient Population
The ECZTEND study was a 5-year open-label extension trial that followed patients who completed one of multiple tralokinumab parent trials (ECZTRA 1–8 and TraSki).9,13,14,20 ECZTEND included 1,672 patients aged ≥12 years with moderate-to-severe AD who received tralokinumab 300 mg every 2 weeks with optional topical corticosteroids or topical calcineurin inhibitors. The median duration of tralokinumab treatment was 2.6 years, and patients were followed for up to 5 years.9,13,14 Of these patients, 1,143 (68.4%) completed treatment, and withdrawals due to lack of efficacy occurred in 7.1% and due to adverse events in 4.3% of patients.20
The post-hoc analysis reported at ISAD included 1,639 patients with available parent trial baseline data.20 These patients had a mean age of 37.2 years on enrolment to the parent trial; 87.5% of patients exhibited head and neck EASI ≥1, and the median head and neck EASI was 2.8. The population had long-standing disease and substantial baseline severity: the mean AD duration was 27.6 years, the median overall EASI score was 27.1, and 47.2% of patients had severe AD (IGA 4).20
Sustained Improvements in Head and Neck Atopic Dermatitis
Long-term findings from the ECZTEND extension trial demonstrated continued improvements in AD with head and neck involvement over the 5‑year follow‑up period.20 Median head and neck EASI decreased from 2.8 at parent trial baseline to 0.4 at ECZTEND trial start (following up to 1 year of treatment in parent trials). The improvement in head and neck EASI was maintained through Week 248 of ECZTEND, with a median head and neck EASI of 0.1 among patients with Week 248 follow-up data (n=84). A sensitivity analysis using last observation carried forward for missing data showed comparable improvement in disease severity, with a median head and neck EASI of 0.2 at Week 248.20
The ECZTEND study showed that improvements in head and neck EASI were comparable to those observed in other body regions.20 When EASI scores were standardised to account for different maximum scale ranges across body regions, the head and neck showed similar patterns of improvement to the upper limbs, lower limbs, and trunk.20
Among patients with head and neck EASI ≥1 at parent trial baseline (n=1,434), the proportion achieving responses increased over time, using various EASI response measures. By Week 248 of ECZTEND, 97.4% (75/77) of those patients had head and neck EASI ≤2, 89.6% (69/77) had head and neck EASI ≤1, 84.4% (65/77) had head and neck EASI-75, and 46.8% (36/77) had complete clearance (head and neck EASI 0).20
Conclusion
The evidence from these studies suggests that tralokinumab can be effective in high-burden areas of moderate-to-severe AD, including the head and neck, hands, and genital regions. Results from the Phase IIIb ADHAND trial suggest that tralokinumab monotherapy is a potentially valuable treatment option for adult patients with moderate-to-severe AD with hand involvement, with 40% of patients achieving clear or almost clear hands at 16 weeks without background topical therapy.12
Real-world data from the TRACE study show that, by 12 months of treatment with tralokinumab, 64.7% of patients with head and neck involvement attained complete or almost complete clearance, with responses observed in both dupilumab‑naive and previously treated patients, and 78% of patients with genital involvement no longer had genital AD.15,18
Long-term data from the ECZTEND extension trial demonstrate that improvements in AD with head and neck involvement are sustained over up to 5 years of tralokinumab treatment.20 The 5-year data showed continued improvement in AD with head and neck involvement, with sub-scores comparable to those observed for other body regions and nearly 90% of patients achieving head and neck EASI ≤1 (in the subgroup of patients with head and neck EASI ≥1 at parent trial baseline) after 5 years of treatment.20
The reported safety profile in the aforementioned trials was consistent with the existing approved label, with low rates of treatment discontinuation due to adverse events, and with no new safety signals reported.12-14,18 Subgroup analysis from the TRACE study suggests that tralokinumab is an effective and well-tolerated treatment for AD in patients who discontinue dupilumab treatment due to conjunctivitis.18 Among adults who discontinued dupilumab due to conjunctivitis, over 80% did not develop conjunctivitis related to tralokinumab therapy in a real-world setting, and tralokinumab treatment was associated with continued improvements in disease severity and quality of life over 12 months.18 When conjunctivitis was reported, it was typically mild-to-moderate and did not lead to treatment discontinuation.18
Collectively, these data contribute to the evidence base supporting the potential role of tralokinumab as a treatment option for patients with moderate-to-severe AD affecting high‑burden anatomical areas, including those who have previously discontinued dupilumab.
Republic of Ireland
Prescribing Information for Adtralza® (tralokinumab) 150 mg solution for injection in pre-filled syringe and Adtralza® (tralokinumab) 300 mg solution for injection in pre-filled pen
Please refer to the full Summary of Product Characteristics (SmPC) (www.medicines.ie) before prescribing.
Indication: Treatment of moderate‑to‑severe atopic dermatitis in adult and adolescent patients 12 years and older who are candidates for systemic therapy.
Active ingredients: Each pre‑filled syringe contains 150 mg of tralokinumab in 1 mL solution (150 mg/mL). Each pre-filled pen contains 300 mg of tralokinumab in 2 mL solution (150 mg/mL).
Dosage and administration: Posology: The recommended dose of tralokinumab for adult and adolescent patients 12 years and older is an initial dose of 600 mg (four 150 mg injections by pre-filled syringe or two 300 mg injections by pre-filled pen) followed by 300 mg (two 150 mg injections by pre-filled syringe or one 300 mg injection by pre-filled pen) administered every other week as subcutaneous injection. Every fourth week dosing may be considered for patients who achieve clear or almost clear skin after 16 weeks of treatment. The probability of maintaining clear or almost clear skin may be lower with every fourth week dosing. Consideration should be given to discontinuing treatment in patients who have shown no response after 16 weeks of treatment. Some patients with initial partial response may subsequently improve further with continued treatment every other week beyond 16 weeks. Tralokinumab can be used with or without topical corticosteroids. The use of topical corticosteroids, when appropriate, may provide an additional effect to the overall efficacy of tralokinumab. Topical calcineurin inhibitors may be used, but should be reserved for problem areas only, such as the face, neck, intertriginous and genital areas. Missed dose: If a dose is missed, the dose should be administered as soon as possible and then dosing should be resumed at the regular scheduled time. Special populations: No dose adjustment is recommended for elderly patients, patients with renal impairment or patients with hepatic impairment. For patients with high body weight (>100 kg), who achieve clear or almost clear skin after 16 weeks of treatment, reducing the dosage to every fourth week might not be appropriate. The safety and efficacy of tralokinumab in children below the age of 12 years have not yet been established. Method of administration: Subcutaneous use. The pre-filled syringe and pen should not be shaken. After removing the pre‑filled syringes or pre-filled pens from the refrigerator, they should be allowed to reach room temperature by waiting for 30 minutes before injecting a pre-filled syringe or 45 minutes before injecting a pre-filled pen. Tralokinumab is administered by subcutaneous injection into the thigh or abdomen, except the 5 cm around the navel. If somebody else administers the injection, the upper arm can also be used. For the initial 600 mg dose, four 150 mg tralokinumab injections or two 300 mg tralokinumab injections, should be administered consecutively in different injection sites within the same body area. It is recommended to rotate the injection site with each dose. Tralokinumab should not be injected into skin that is tender, damaged or has bruises or scars. A patient may self‑inject tralokinumab or the patient’s caregiver may administer tralokinumab if their healthcare professional determines that this is appropriate.
Contraindications: Hypersensitivity to the active substance or to any of the excipients.
Precautions and special warnings: If a systemic hypersensitivity reaction (immediate or delayed) occurs, administration of tralokinumab should be discontinued and appropriate therapy initiated. Patients treated with tralokinumab who develop conjunctivitis that does not resolve following standard treatment should undergo ophthalmological examination. Patients with pre‑existing helminth infections should be treated before initiating treatment with tralokinumab. If patients become infected while receiving tralokinumab and do not respond to antihelminth treatment, treatment with tralokinumab should be discontinued until infection resolves. Live and live attenuated vaccines should not be given concurrently with tralokinumab. Adtralza contains polysorbate 80 (E 433) as an excipient, which may cause allergic reactions.
Interactions: See SmPC for full details on interactions.
Fertility, pregnancy and lactation: There is limited data from the use of tralokinumab in pregnant women. Animal studies do not indicate direct or indirect harmful effects with respect to reproductive toxicity. As a precautionary measure, it is preferable to avoid the use of tralokinumab during pregnancy. It is unknown whether tralokinumab is excreted in human milk or absorbed systemically after ingestion. Animal studies did not show any effects on male and female reproductive organs and on sperm count, motility and morphology.
Side effects: Very common (≥1/10): Upper respiratory tract infections. Common (≥1/100 to <1/10): conjunctivitis, conjunctivitis allergic, eosinophilia, injection site reaction. Uncommon (≥ 1/1 000 to < 1/100): Keratitis.
Precautions for storage: Store in a refrigerator (2°C‑8°C). Do not freeze. Store in the original package in order to protect from light.
Legal category: POM.
Marketing authorisation number and holder: Pre-filled syringe: EU/1/21/1554/002. Pre-filled pen:
EU/1/21/1554/004.
LEO Pharma A/S, Ballerup, Denmark.
Last revised: February 2026.
Reference number: MAT-91356.
Further information can be found in the Summary of Product Characteristics or from: LEO Pharma, Cashel Road, Dublin 12, Ireland. e-mail: [email protected]
® Registered trademark
| Reporting of Suspected Adverse Reactions
Adverse events should be reported. For the United Kingdom, reporting forms and information can be found at: yellowcard.mhra.gov.uk. Adverse events should also be reported to Drug Safety at LEO Pharma by calling +44 (0)1844 347333 or e-mail [email protected] or search for MHRA Yellow Card in the Google Play or Apple App Store. For the Republic of Ireland, reporting forms and information can be obtained from HPRA Pharmacovigilance, Tel: +353 1 6764971, Website: www.hpra.ie, E-mail: [email protected]. Adverse events should also be reported to Drug Safety at LEO Pharma by calling +353 1 4908924 or e-mail: [email protected]. |
MAT-91753 May 2026